Recent consensus advice on integrated guidance to enhance the care of patients with familial hypercholesterolaemia (FH) in Australia provides a timely reminder and opportunity for general practitioners (GPs) to increase their awareness and skill in diagnosing and managing the condition.1
With 88% of Australians presenting to their GPs annually,2 GPs are well placed to play a more active part. Currently, fewer than 10% of Australia’s 100,000 patients with FH are diagnosed, with most failing to achieve optimum management and therapeutic targets.3
The hereditary nature of FH, with its high cholesterol burden present from birth,3 causes premature atherosclerotic cardiovascular disease (ASCVD), principally coronary artery disease (CAD), and death if left untreated.4,5 FH is caused by a defect in the low-density lipoprotein (LDL) receptor pathway, and it affects one in 250-300 Australians.6,7 Among first-degree relatives, 50% are affected because of its autosomal dominant, monogenic inheritance and high penetrance.3
General practice is central to the continuity of care and advocacy for all patients with FH and their families. This involves a role in screening, diagnosis, shared care with specialists, overseeing cholesterol-lowering medications and multimorbidities, as well as applying context-specific models of care for FH.1,8
This article focuses on key recommendations for the identification and management of heterozygous FH derived from the recently published integrated guidance,1 with particular reference to GPs. The aim of this article is to help translate this updated guidance into everyday health policy and practice, and facilitate the provision of high-quality healthcare for patients with FH and their families in the primary care setting.
New genetic tests and impact on clinical care
The Australian Government has introduced new pathology services to assist with detection of heritable mutations predisposing to FH: Medicare Benefits Schedule (MBS) Item 73352 for index cases and Item 73353 for cascade testing of close relatives.9 Item 73352 requires non-GP specialist authorisation, but Item 73353 can be requested by the patient’s GP.
These new tests provide diagnostic precision, offering GPs added incentive to increase diagnosis in the young when treatment can be most effective.4,9 The level of evidence is high, and the class of recommendation is strong for using genetic testing to confirm the diagnosis. This is especially important when cascade testing is planned.1,10,11
Since more than 20% of probable or definite FH may not have a detectable mutation,11 FH should not be totally excluded if a pathogenic, or likely pathogenic, gene variant is not detected.1,4,12
A shared-care approach between GP and non-GP specialists (ie lipidologist, cardiologist, pediatrician)13 can facilitate genetic testing of index cases. Pre- and post-test counselling should be an integral part of the process.1 Once an index case is genetically proven, GPs can offer cascade testing among first- and second-degree relatives, arrange appropriate counselling and collaborate with non-GP specialists in risk stratification and treatment.8,12 As discussed later, screening requires appropriate GP education, screening tools and skill training in the care of patients with FH.
Figure 1 summarises genetic screening and management of an individual at high risk of FH.
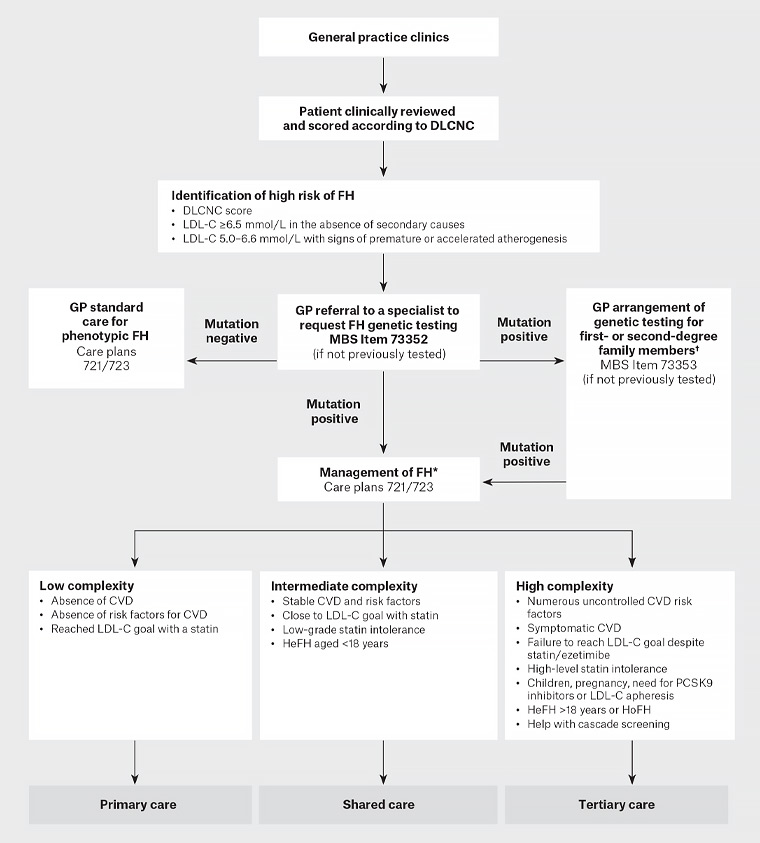
Figure 1. Process for genetic screening and management of an individual at high risk of familial hypercholesterolaemia. Click here to enlarge
*Refer to Sturm AC, Knowles JW, Gidding SS, et al, Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel, J Am Coll Cardiol 2018;72(6):662–80. doi: 10.1016/j.jacc.2018.05.044.
†Genetic cascade testing may be undertaken by a general practitioner with skills in the care of patients and families with FH, under the guidance of an appropriate specialist. Consent is obtained from the index case to contact family. The process of risk notification of family members should be consistent with relevant local legislation and institutional guidelines. Risk notification may be indirect (providing a family letter for the notifier to pass to relatives) or direct (clinical service writes to relatives); pre-and post-test genetic counselling should be offered to all at-risk family members.1
CVD, cardiovascular disease; DLCNC, Dutch Lipid Network Criteria; FH, familial hypercholesterolaemia; GP, general practitioner; HeFH, heterozygous familial hypercholesterolaemia; HoHF, homozygous familial hypercholesterolaemia; LDL-C, low-density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin 9
Risk notification of family members requires special skills to overcome potential barriers, such as privacy laws, poor communication in families, poor health literacy, geographical location and psychological issues.8,11 Coordination of the overall process remains a significant challenge, especially from current inadequate general practice infrastructure.1,8,14
Diagnosis in adults
Awareness of FH among Australian GPs
Despite the high prevalence of FH and an ever-increasing number of research publications, awareness about FH in the general community and among many health professionals remains suboptimal.3 The concept of increased cholesterol burden from birth in affected individuals is not well understood. In addition, the hereditary ‘familial’ component of FH, with its associated premature ASCVD, tends to remain unrecognised and untreated.3
Young people have most to gain from early diagnosis and appropriate treatment.4,15,16 Apart from lifestyle interventions, most patients will need lifelong cholesterol-lowering medications, especially statins. Ezetimibe and proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors may be needed for cases that are more difficult to control.3,4,17
Failure to diagnose FH before middle age results in up to 50% of untreated men developing myocardial infarction or angina by the age of 50 years, and 30% of untreated women similarly affected by the age of 60 years.5 Early recognition and treatment produces significant reductions in cardiac morbidity and mortality.15,16 Strict adherence to treatment allows affected individuals to live a normal lifespan.3
History-taking
The importance of effective history-taking in clinical medicine is one of the fundamental tenets of diagnostic evaluation. However, poor-quality medical information in practice records may affect the diagnosis. GPs will often be aware of patients and families with a history of premature coronary events attending their practice. Linking such histories, combined with a lipid profile showing total cholesterol >7.5 mmol/L or LDL cholesterol (LDL-C) >5.0 mmol/L, should alert the GP to the possibility of FH. Potential secondary causes of hypercholesterolaemia (diabetes, hypothyroidism, steroid use, renal and liver disease) should be excluded before FH is further assessed.3
Personal or family history of premature CAD and markedly elevated cholesterol levels, both central to the Dutch Lipid Clinic Network Criteria (DLCNC) score, are the chief drivers for establishing a phenotypic FH diagnosis. Assessment of non-cholesterol risk factors, genetic testing and cardiovascular imaging can improve risk re-stratification and optimise treatment.3
Electronic health record screening
FH meets all the criteria for worthwhile disease screening.8 Various approaches have been suggested – including universal, opportunistic and selective screening – but all require effective coordination for maximum benefit.8,10
Reverse child–parent screening,18 targeting premature myocardial infarcts in coronary care units and screening electronic health records (EHRs) in general practice all offer promise.8,19,20 Laboratory report alerts also help with index case detection.1,8,20
Purposeful clinical examination
Once FH diagnosis is considered, clinical examination should focus on physical stigmata including premature corneal arcus and tendon xanthomata.3 Periorbital xanthelasmata are suggestive of hypercholesterolaemia, and their presence should alert the GP to the potential of underlying FH. Additional training for GPs in pattern recognition and early consideration of FH among close family members could help increase awareness of key hereditary features.
The DLCNC score combines personal and family history of premature CAD,1,3,8 elevated LDL-C levels and physical stigmata to establish phenotypic FH diagnosis in high-risk patients. In a genetic analysis of FH in Australia, 70% of patients with a definite FH diagnosis were found to have an FH-causing mutation, but only 29% of patients with probable FH and 11% of patients with possible FH were mutation positive.21
Diagnosis in children and adolescents
The cumulative LDL-C burden in patients with FH starts from birth and progressively increases over their lifetimes.4 However, most children with FH are asymptomatic and have no clinical signs. Hence, the current largest gap is the early detection of children with FH.3
Early detection is an important component of providing optimum management, most likely statins, from the age of 8–10 years.1 Use of statins in children will need parental consent with adequate explanation of its benefit and possible side effects. The benefits outweigh any risks for most children.
The LDL-C level drives the phenotypic diagnosis, but genetic cascade testing offers the definitive diagnosis.4,11 Risk stratification helps to facilitate more rational and precise treatment.8
The DLCNC score is not suitable for use in children and adolescents.3 Children with an LDL-C ≥5 mmol/L have a high risk of FH; for children with premature CAD in close relatives and/or baseline high cholesterol in one parent, an LDL-C ≥4 mmol/L is indicative of high FH risk.4
The current guidelines do not recommend specific cut-off LDL-C levels for parental hypercholesterolaemia.1,4 The value of non-invasive cardiovascular imaging tools, such as the carotid intima-media thickness test to improve the early diagnosis of FH in children, needs further evaluation.
Management
Adults
There is compelling evidence from extensive clinical trial, registry and genetic data for patients with FH to be actively treated with cholesterol-lowering medication, diet and lifestyle modification from an early age.
8,12 Other risk factors should also be addressed. Shared doctor–patient decision making helps adherence to therapy.
22
Cumulative evidence supports a 50% reduction in LDL-C, plus low absolute targets depending on primary or secondary prevention settings.8,23,24 Such targets are rarely achieved, with evidence from a Danish study showing less than 50% adherence to treatment even with an established FH diagnosis,25 consistent with a recent FH registry report in Australia.26 Preliminary evidence from an Australian primary care study shows a similar pattern.27
To reach very low LDL-C levels, patients may require sequential treatment with a high-potency statin, ezetimibe and PCSK9 inhibitor,8,22,23 the latter now listed on the Pharmaceutical Benefits Scheme (PBS) requiring initiation by a non-GP specialist.28 Follow-up consultations can be managed in primary care for patients with low-complexity FH. Greater awareness by Australian GPs would help optimise shared-care responsibility with specialists in multiple disciplines for patients at greatest risk.8
FH treatment from childhood is supported by good-quality observational studies.4,8 Modest, sustained reductions in LDL-C starting early in life can have a major effect in preventing future premature mortality due to ASCVD.4,8,10,18 A healthy lifestyle, while important, does not sufficiently lower LDL-C.
A low-potency statin, with or without ezetimibe, may be required from the age of 10 years.4,8,10,15,29,30 LDL-C targets do not need to be as low as for adults,8,16 but medication safety should be continually monitored.4,8
Family-based clinics with paediatric specialist involvement that address therapy adherence are essential for enhancing care.1,8 If uncertainty exists and GPs feel reluctant to initiate or continue statin treatment, support from a paediatric specialist with expertise in lipidology should be sought.1,3
Box 1 summarises recommendations and therapeutic targets, based on moderate levels of evidence and class of recommendation, for managing FH.1,24,26,29,30
| Box 1. Low-density lipoprotein cholesterol treatment targets and recommendations for the management of familial hypercholesterolaemia |
Adults
- Commencement of statin treatment should be considered once a diagnosis of FH is confirmed.1
- LDL-C targets can be divided as follows:
- LDL-C <2.5 mmol/L (absence of ASCVD or other major ASCVD risk factors)1,20,21
- LDL-C <1.8 mmol/L (imaging evidence of ASCVD alone or other major ASCVD risk factors)1,20,21
- LDL-C <1.4 mmol/L (presence of clinical ASCVD).1,20,21
Children
- Commencement of statin treatment should be considered by age 8–10 years irrespective of sex.1,4,6,8,13,20,27
- For children with FH aged 8–10 years on a suitable diet, an LDL-C treatment target <4.0 mmol/L or a 30–40% reduction in LDL-C may be acceptable.1,4,6,13,27
- In children aged >10 years who are maintained on a suitable diet, an LDL-C treatment target <3.5 mmol/L or a 50% reduction in LDL-C may be acceptable.1,4,6,13,27
- Statin therapy with or without ezetimibe in addition to diet/exercise should be used to reach the above targets.1,4,6,8,13,27
- Statins licensed for use in children aged 8–10 years (pravastatin, fluvastatin, simvastatin) should be used; ezetimibe is licensed from the age of 10 years and should be used accordingly.1,2
- Atorvastatin and rosuvastatin use should be considered according to the clinical indications and shared decision making.1,6
- PCSK9 inhibitors are not generally recommended in children but may be considered in heterozygous FH according to clinical indications and shared decision making. Further clinical trials on long-term safety are required.
|
| ASCVD, atherosclerotic cardiovascular disease; FH, familial hypercholesterolaemia; LDL-C, low-density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin 9 |
Pregnancy
Statins and other systemically absorbed cholesterol-lowering medications should be ceased three months before planned conception as well as during pregnancy and while breastfeeding.1 All women of childbearing age with FH should be offered pre-pregnancy counselling prior to starting treatment with statins. Appropriate contraceptive advice that is reinforced at least annually helps minimise cardiovascular risk. GPs should always seek specialist support for patients with FH who are considering pregnancy.3
The importance of early diagnosis and treatment of FH in girls should never be underestimated.31 Pregnancy and lactation can result in the loss of effective years of statin treatment due to childbearing.31 Adherence to statin treatment can be difficult, especially in young people. The need for early and ongoing treatment at optimal doses should be stressed to improve future ASCVD outcomes.31 The cost-effectiveness of such an approach is increasingly recognised.32
Conclusion
This guidance is aligned with a recent international global call to action on FH.33 The recommendations are designed to be incorporated into healthcare pathways that meet the needs of the Australian population.3,8 Several efforts have been made to improve care of patients with FH in primary care. These include increased recognition of phenotypic diagnosis of FH, greater consideration of screening in children, increased awareness of the need to employ implementation science and practice to optimise health service delivery, and advocacy groups. The introduction of MBS items for genetic testing and PBS-supported use of PCSK9 inhibitors is likely to improve considerably the care of patients with FH in the future.9,12,28 Education and skill training for GPs, such as cascade screening, risk notification, and pre- and post-test genetic counselling, are paramount for implementing effective care in general practice.
The challenge we now face is how to implement this guidance into health policy and high-quality care. Implementation research and practice will need to be embraced as a high priority to increase the impact of this guidance on improving the care of all Australians who have, or are at risk of, FH.33
Key points
- Recent guidelines support improved care of patients with FH in the primary care setting.
- GPs are ideally placed to play a proactive part in diagnosing FH and then co-managing these patients in conjunction with other specialists.
- New MBS and PBS items enable more precise diagnosis and treatment of FH.