Creutzfeldt–Jakob disease (CJD) was first described by neurologists Creutzfeldt and Jakob in the 1920s, with the modern interpretation made by Miller Fisher in 1960.1 It is a rare, uniformly fatal neurodegenerative disease manifested by rapidly progressive dementia, myoclonus, ataxia, visual disturbances, extrapyramidal and pyramidal involvement, and kinetic mutism.2,3 There are several hypotheses regarding the mechanism underlying CJD, which often involves prions that can convert into insoluble aggregates in nervous tissue4 and result in neuronal loss, astrocytic proliferation, spongiform change and abnormal disease-related, protease-resistant prion protein (PrP) in tissues.5
Sporadic CJD (sCJD) is the most common form6 of CJD (85–90%). The mean age for onset of symptoms is 65 years, with a range of 41–91 years.7 The average duration of symptoms is four months.8
Diagnostically, CJD is a challenging condition to detect premortem because of non-specific clinical manifestations. Definitive diagnosis requires identification of PRPCJD, the pathological form of PRP, in a brain biopsy.9 Suggestive investigation with cerebrospinal fluid (CSF) protein 14-3-3 has a sensitivity and specificity of 92% and 80% respectively. Magnetic resonance imaging (MRI), either diffusion-weighted MRI or fluid attenuation inversion recovery (FLAIR), features have 91–92.3% and 93.8–95% sensitivity and specificity respectively.10 Electroencephalography (EEG) showing patterns typical of CJD has a reported 64% sensitivity and 91% specificity for diagnosis.11
Diagnostic criteria for a ‘definite’ case of sCJD described by the World Health Organization (WHO) requires neuropathological confirmation by brain biopsy and/or identification of PRPCJD (ie immunocytochemistry, western blot) and/or presence of scrapie-associated fibrils.12 It should be noted, however, that the WHO does not recommend brain biopsy for the diagnosis of suspected CJD.13 There is no consensus about a diagnostic pathway. Manix et al suggested in 2015 that a diagnostic pathway should require presentation of rapidly progressive dementia, clinical signs, suggestive investigation findings for CJD, and routine investigations negative for other diagnoses.14
Case
RE, aged 67 years, lived on, and managed, a large sheep farm with his brother for 50 years. He presented with two to three months of rapid decline in cognition and function. RE was previously independent, with all activities of daily living, and worked full time.
RE was disoriented and could not provide much history. Collateral histories from his brother and his general practitioner (GP) revealed a rapidly progressive impairment in orientation, managing paperwork and unintentional weight loss over three months. He had no significant past medical history and took no regular medication. RE spent his entire life in a rural area of South Australia and did not travel overseas. He has no known family history of dementia.
On examination, RE was disorientated to place, time and person. He had optic ataxia, demonstrated by the inability to move his hands towards objects within his range of vision, and left-side visual inattention. Readers should note that this clinical feature differs from cerebellar ataxia in that the patient will be able to perform tasks smoothly once given proprioceptive or auditory cues. Features reported to present frequently in CJD, such as myoclonus, pyramidal or extrapyramidal signs, and abnormalities of gait were not present.
Autoimmune screening, vasculitic screening, thyroid peroxidase antibodies, thyroid function tests, syphilis serology, human immunodeficiency virus serology, Epstein–Barr virus and hepatitis serologies were all negative. Full blood count, urea, creatinine and liver function tests were all in normal ranges. A computed tomography scan of the brain showed no features suggesting an alternative diagnosis. An MRI of the brain showed features of high‑signal cortical ribboning, prominent at marked areas on FLAIR images (Figure 1A), correlated with diffusion restriction on diffusion-weighted images (Figure 1B). Classically, in CJD, an increased T2 signal is observed in the head of the caudate. However, a widespread increase in cerebral cortical signal has been observed in CJD, particularly associated with specific subtypes.15 The prominent parieto-occipital cerebral high signal may be seen as consistent with optic ataxia, which is classically associated with parietal pathologies.
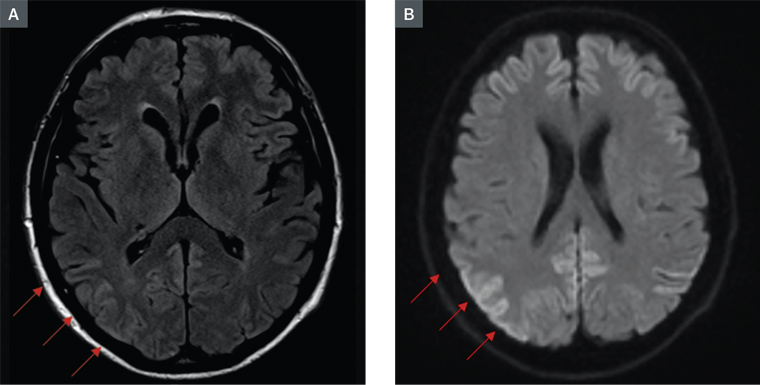
Figure 1. Magnetic resonance imaging of RE's brain. A. FLAIR image; B. Diffusion-weighted image
On EEG, quasi-periodic discharges were seen following hyperventilation. There was no periodic sharp wave pattern typical of CJD. The CSF had an elevated protein level of 0.9 g/L (reference range 0.1–0.65 g/L) with no cells. The level of CSF protein 14-3-3, a protein marking rapid neuronal destruction and suggesting CJD, was later found to be elevated.
A diagnosis of probable sCJD was made. RE’s brother, being the only family available, was counselled regarding the grave prognosis of CJD and advised that it would become increasingly difficult to manage RE at home. RE was discharged from hospital to an aged-care facility for palliative care. He continued to decline and, unfortunately, died three weeks following discharge.
Discussion
CJD is a rare cause of neurodegenerative disease, with an incidence of one in one million per year.16 The case described in this article was a presentation of CJD that had a few of the key physical findings for this condition. The case highlights the need for awareness of the disease in an ageing Australian population, given that many people present to primary care practice with cognitive impairment. Primary care providers should make an early referral to tertiary centres for early work-up for patients with acute cognitive decline lacking a clear cause.
A critical aspect of this case was the exposure history. To the best of our knowledge, no author has clearly demonstrated that exposure to sheep is associated with sCJD. This association was previously refuted in a 2008 study by Georgsson et al17 but, interestingly, other authors (eg Cassard et al) demonstrated in 2014 that sheep scrapie isolates could result in the emergence of prions with a similar phenotype as that associated with sCJD in mice models.18
RE’s brother, who has the same exposure and lifestyle, appears to have symptoms of early cognitive impairment, which may hint at the same presentation. He was not formally assessed for possible CJD. We have asked the Australian National CJD Registry to follow-up on the case and asked RE's brother for consent to be followed up regarding this. Interventions that may be helpful involve regular monitoring for progression of symptoms and early diagnostic work-up, should suggestive clinical features present.