Differences/disorders of sex development (DSD) or ‘intersex’ encompass a wide range of conditions usually presenting in the newborn period or adolescence. Most are relatively rare, and the overall incidence depends on which conditions are included; for example:
- Klinefelter syndrome (47,XXY) – one in 450 births of male infants1
- discordance between phenotypic and genotypic sex at birth – one in 4500–5500 births2
- congenital adrenal hyperplasia (CAH) – one in 14,000 births3
- complete XX or XY sex reversal – one in 20,000 births2
- ovotesticular DSD – one in 100,000 births.2
Terminology has been a source of dissatisfaction among many affected individuals, with some peer support groups having a strong preference for ‘intersex’ over ‘DSD’,4 while others actively dissociate from the term ‘intersex’. Clinicians generally avoid either term when speaking with affected individuals and their families, choosing to use terminology based on the diagnosis whenever possible (eg CAH, Klinefelter syndrome). However, DSD/intersex can be useful umbrella terms in the context of medical literature and policy. The diverse clinical features, pathophysiology and management challenges in DSD have compounded efforts to reach consensus about nomenclature and optimal management. Promoting a non-binary view of sex and gender can be helpful to work towards common goals of optimal care. There is limited evidence to support and/or guide clinical decision making that involves the consideration of medical, human rights, ethical and legal aspects.5 Moreover, there has been considerable debate regarding the indication and timing of surgical interventions for minors with DSD.
Clinical features and underlying pathophysiology vary from one individual with a DSD to another. As a result, presentations can range from atypical genitalia in infancy to atypical pubertal development in adolescence to fertility concerns in adulthood. Throughout the lifespan, there can be challenging and sensitive discussions and decisions, such as the appropriate sex of rearing, gender identity, potential for future adult sexual functioning and fertility. Healthcare professionals may face pressure for urgent diagnosis and management at presentation but also need to consider a medium-term and long-term plan with the child/adolescent and family in the context of the social and cultural environment. Psychological support should be provided throughout the patient journey.
As a result of the rarity of many conditions, data on long-term outcomes are limited, and patient groups as well as professionals have challenged aspects of clinical care that were previously considered as standard. Through greater use of multidisciplinary teams (MDTs), development of patient registries and international collaboration, the evidence base for the best standards of care will continue to develop.
Classification and diagnosis of DSD
The development of the 2006 consensus guidelines6 was a turning point in the classification of DSD, with three broad categories: sex chromosomal DSD, 46,XX DSD and 46,XY DSD (Box 1). This resulted in the abandonment of misleading and pejorative terms such as ‘hermaphrodite’. Diagnosis of DSD generally involves a hormone profile work-up, genetic testing and visualisation of internal reproductive structures, through either medical imaging or laparoscopy. These investigations are coordinated by a member of the MDT, often the paediatric endocrinologist.7 Throughout the diagnostic work-up and discussion of results and management plans, it is paramount that there is an open and transparent discussion with parents and affected individuals at developmentally appropriate times.
| Box 1. Summary of classification of differences/disorders of sex development taken from the 2006 consensus statement6 |
46,XY DSD
- Disorders of testicular development, including partial and complete testicular dysgenesis, testicular regression and ovotesticular DSD
- Disorders of androgen biosynthesis (eg 5-alpha reductase deficiency) or action (CAIS, PAIS, luteinising hormone receptor defects)
- AMH disorders (eg persistent Müllerian duct syndrome)
- Other disorders, including severe hypospadias and cloacal exstrophy
46,XX DSD
- Disorders of ovarian development, including ovarian dysgenesis as well as testicular or ovotesticular DSD
- Disorders of androgen excess of either fetal, fetoplacental or maternal origin
- Other disorders, including cloacal exstrophy, MURCS and vaginal atresia
Sex chromosome DSD
Disorders related to atypical arrangement of sex chromosomes, including:
- missing sex chromosome (eg 45,X, as seen with Turner syndrome)
- additional sex chromosome (eg 47,XXY, as seen with Klinefelter syndrome)
- more than one type of sex chromosome arrangement (eg 45,X/46,XY or 46,XX/46,XY)
|
| AMH, anti-Müllerian hormone; CAIS, complete androgen insensitivity syndrome; DSD, differences/disorders of sex development; MURCS, Müllerian, renal, cervicothoracic somite abnormalities; PAIS, partial androgen insensitivity syndrome |
Neonates
Neonates who require diagnostic work-up include those with ambiguous or atypical genitalia or less obvious findings such as bilateral nonpalpable gonads, palpable gonads in labioscrotal folds, perineal hypospadias, undervirilised phallus, clitoromegaly or discordant genital appearance to prenatal genetic test. The initial evaluation should include a thorough pregnancy and family history and clinical examination. Physical examination should include evaluation of general health, hydration state, blood pressure and appearance of external genitalia with regards to whether the labioscrotal folds are fused, the presence of palpable gonads, the location of the urethral meatus and the size of the phallus/clitoris.8 Palpable gonads are most commonly testes and occasionally ovotestis as ovaries tend to remain in pelvic position.9 DSD may also be part of a multisystem disorder with coexistent metabolic abnormalities or dysmorphic features. CAH is the most common cause of genital ambiguity in infancy and should be considered in any baby with genital ambiguity, shock and/or hyponatraemia and hyperkalaemia.10 It is a potentially life-threatening condition and requires urgent treatment with high-dose hydrocortisone to prevent an adrenal crisis.
Initial investigations for genital ambiguity include karyotype and/or rapid polymerase chain reaction or fluorescence in situ hybridisation for Y material, glucose, electrolytes, 17-hydroxyprogesterone (17-OHP), gonadotropins, oestradiol and testosterone. Results should be interpreted in the context of the baby’s gestational and chronological age. The paediatric endocrinologist may request further testing including pelvic ultrasonography, urine steroid profile, serum anti-Müllerian hormone, human chorionic gonadotropin stimulation test and DSD gene panels.
Sex assignment is a dilemma in a small percentage of patients with DSD and requires an individualised approach taking into consideration prenatal androgen exposure, fertility potential, quality of sexual function, surgical options, gonadal pathology/malignancy risk and potential adult gender identity.2 Given the variation and complexity of different types of DSD, it may be difficult to extrapolate outcomes for each individual. Research in DSD and gender identity has only been done in larger subgroups such as patients with CAH and androgen insensitivity syndrome. To date, evidence suggests sex of rearing is an important but imperfect predictor of gender identity, with some additional contribution of prenatal androgen exposure in male gender identity.11
Children and adolescents
Simple virilising CAH may present in childhood with a rapid growth spurt, exaggerated signs of adrenarche (eg pubic hair) and virilisation of the genitalia, with clitoromegaly or penile growth without testicular enlargement.10 A 17-OHP level, synacthen test and genotyping are useful investigations for diagnosis.
Presenting features of DSD in an adolescent may include delayed puberty, primary amenorrhea or virilisation of a phenotypic female.10 Non-classical CAH may have similar features to polycystic ovarian syndrome. Physical examination of the external genitalia may be best deferred and only performed by an experienced urologist or gynaecologist under an anaesthetic. Investigations include karyotype, serum electrolytes, pubertal hormone profile, androgens, pituitary hormone profile, adrenal steroid profile and transabdominal pelvic ultrasonography. Magnetic resonance imaging and ultrasonography of internal structures can be inaccurate, and internal structures ‘not visualised’ does not equate to absent structures or lack of future function (eg a small uterus may be due to lack of oestrogen effect). It may only be at genitoscopy and laparoscopy that internal structures are confidently identified. The results from these investigations need careful interpretation in the context of other clinical features. For example, an adolescent with primary amenorrhoea who has normal breast development could have complete androgen insensitivity syndrome (CAIS) with a 46,XY karyotype, or Müllerian agenesis, also known as Mayer–Rokitansky–Küster–Hauser syndrome, with a 46,XX karyotype. An adolescent with primary amenorrhoea and little/no breast development and short stature could have Turner syndrome, while one with normal stature could have complete gonadal dysgenesis.
Genetic testing
Many DSD are caused by variants in genes involved in genital and/or gonadal development or steroid hormone production. Recent advances in molecular genetics have enabled more accurate molecular diagnoses in many individuals, which can then provide guidance on prognosis and potential gonadal malignancy risk for some (eg CAIS).12 Furthermore, a genetic diagnosis is often helpful for individuals and families as they deal with uncertainty, potential stigma and blame. The yield from diagnostic gene testing varies from high in cases with a clear steroidogenic pathway defect (eg CAH) to historically low in 46,XY DSD. Targeted gene panels have improved the diagnostic yield to almost 50% of 46,XY DSD.13 The reducing cost and greater accessibility of whole-exome sequencing (WES) and whole-genome sequencing (WGS) may further increase the diagnostic yield of genetic testing. It is paramount that appropriate genetic counselling be undertaken for all genetic testing and especially for potential unexpected findings from WES and WGS.
Management
Multidisciplinary teams
A multidisciplinary approach to shared decision making is considered the ‘crux of patient-centred care’14 and involves simultaneous, but independent, contributions of team members from two or more disciplines.2 The core composition of the team may vary according to the age of the child, DSD type and local resources, and may include a representative from endocrinology, urology, gynaecology, genetics, psychology, ethics and/or hospital executive (Figure 1). With the patient’s/parent’s consent to share information outside the hospital setting, communication with the family’s primary care physician is important to nourish the therapeutic relationship in primary care, enable ongoing education of health professionals and provide a smoother transition to adult care.
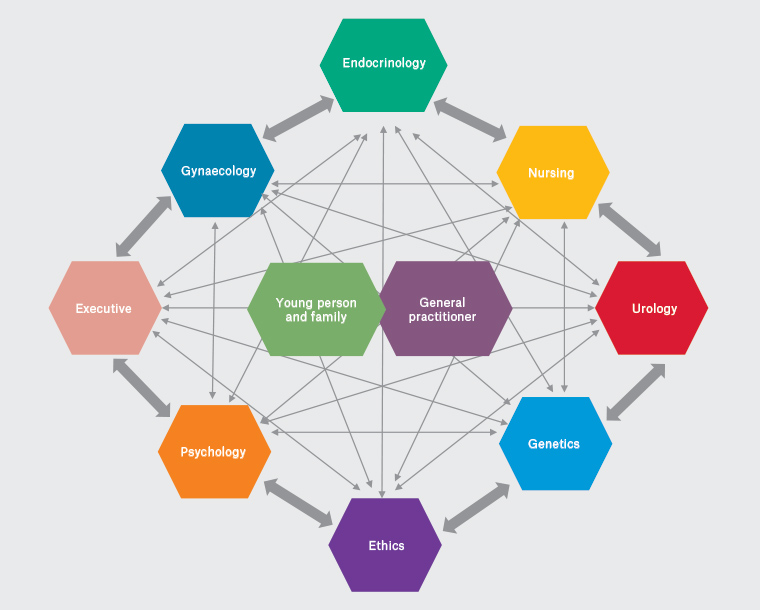
Figure 1. Multidisciplinary team model2
In some centres, prospective, systematic peer review of complex decision making for patients with DSD has been formalised through regular multidisciplinary meetings. An audit of the DSD MDT meetings in Sydney (The Children’s Hospital at Westmead, Sydney Children’s Hospital) and Melbourne (Royal Children’s Hospital, Monash Children’s Hospital)15 highlighted a number of areas discussed at the meetings (Box 2). The audit also showed that an active deferral of surgery was the outcome in >20% of cases, surgery was only recommended for therapeutic indications, and >90% of the gonadectomies performed had abnormal features on histology.15
| Box 2. Common issues discussed at multidisciplinary team meetings |
- Complex differences/disorders of sex development cases with diagnostic dilemmas
- Sex of rearing
- Timing and/or indication for genital tract surgeries
- Timing and/or indication for hormone treatment
- Risk of gonadoblastoma
- Timing and/or indication for gonadectomy
- Discordance between the parents’ view and the members of the multidisciplinary team, conflicts of opinion within the team
- Psychosocial wellbeing of the young person and family
- Referral to peer support groups
|
Psychological support
Parents of newborns with DSD should be provided with psychological support, ideally by someone with experience in DSD. The clinical psychologist is able to provide support at times of medical/surgical discussions and interventions, gender identity assessments of the young person, and transition to adult services. The psychologist can aid in assessing the parents’ and young person’s understanding of information discussed and provide family support in a culturally sensitive manner. The psychologist can also liaise with the general practitioner and guide individuals and families to appropriate peer support groups and resources. Unfortunately, access to psychological support is limited in Australia and around the world,16 and these roles may be undertaken to a variable degree by doctors, nurses, genetics counsellors and other health team members. Parent and peer support groups can be helpful for many families (Table 1).
It is important that any information disclosure is done in a sensitive manner, with language that is appropriate for the patient’s developmental age and parental capacity to understand.17 There may be uncertainty about outcomes, and parents and patients may require repeated explanation about the condition because information retention can be an issue, especially during the stressful period of a new diagnosis. It is also important to bear in mind that the focus of explanation and the issues of discussion change over the time. Information disclosure can present further challenges for culturally and linguistically diverse patients, as concepts about sexuality and gender roles may vary between different cultures.18 A systematic approach to medical record keeping also helps with future disclosure of the basis on which management decisions were made.
| Table 1. List of support groups and online resources for parents and health professionals |
| General information about differences/disorders of sex development (DSD) |
dsdfamilies – information and support for families, www.dsdfamilies.org
dsdteens – information for young people with DSD, www.dsdteens.org
DSD genetics, http://dsdgenetics.org/index.php |
| Patient advocacy/human rights |
Intersex Human Rights Australia (IHRA), https://ihra.org.au |
| Congenital adrenal hyperplasia (CAH) |
Congenital Adrenal Hyperplasia Support Group Australia (CAHSGA), www.cah.org.au
Australasia Paediatric Endocrine Group, https://apeg.org.au/patient-resources/hormones-me-booklet-series
CARES Foundation (USA based), www.caresfoundation.org
CAH IS US (UK based), www.cahisus.co.uk/index.htm |
| Androgen insensitivity syndrome (AIS) and/or other intersex/DSD conditions |
Intersex Peer Support Australia (IPSA), https://isupport.org.au
AIS-DSD Support Group (USA based), http://aisdsd.org
Accord Alliance, www.accordalliance.org |
| Turner syndrome |
Turner Syndrome Association of Australia, www.turnersyndrome.org.au |
| Klinefelter syndrome and variants |
Australian X & Y Spectrum Support (AXYS), https://genetic.org
Klinefelter Syndrome Australia Inc., https://klinefelter.org.au
XYY Syndrome Association of Australia Inc., https://xyyaustralia.org
Triple X & Trisomy X Support Group Australia (closed Facebook support group),
www.facebook.com/groups/TrXAUS |
| Hypospadias, bladder/cloacal exstrophy |
Bladder Exstrophy, Epispadias, Cloacal Exstrophy, Hypospadias, Australian Community (BEECHAC), www.beechac.com/about-us.html |
| Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome/Müllerian agenesis |
Sisters for Love MRKH Foundation, www.sistersforlove.org/home.html |
Surgical decisions
Surgical decisions remain the most challenging and controversial area in DSD, especially if the surgery is considered irreversible. Functionality, malignancy prevention and cosmetic appearance are all taken into account when considering surgical procedures.19 There is currently no consensus in relation to the need for, or optimal timing of, many surgical interventions. Over time, recommendations for surgery have evolved for reasons including:
- a cessation of the standard practice to remove gonads solely to align with sex of rearing
- advances in immunohistochemistry and genetics that have improved tumour risk stratification
- patient advocate requests for deferral of non-therapeutic surgery, despite a lack of universal interpretation of ‘medically necessary surgery’.
There is a paucity of longitudinal and systematic studies on the impact of surgery versus deferral of surgery to guide optimal management. DSD MDT meeting reviews consider these factors and may result in active deferral of surgical intervention. Concerns about the appropriateness of surgery were the subject of an Australian Senate Inquiry in 201320 and a recent Australian Human Rights Commission review on protecting the human rights of people born with variations in sex characteristics (report awaited).
Predicting the risk of germ cell tumour (GCT) development in DSD is an area of ongoing research and varies depending on the presence of gonadoblastoma on Y chromosome (GBY) material in the karyotype, anatomical localisation and degree of maturation of the gonad. The highest risk is in patients with undifferentiated gonadal tissue, dysgenetic testes and intra-abdominal testes in undervirilisation syndromes.21,22 Gonadoblastoma and invasive GCTs occur at a much younger age when compared with germ cell neoplasia in situ, which occurs mainly after the exposure to puberty hormones.21
Delaying surgery for functional gonads allows time for exploration of gender identity, which may differ from assigned sex; however, it is important to bear in mind the lack of robust surveillance methods to predict malignancy risk if dysgenetic gonads are left in situ. The use of gonadotropin-releasing hormone agonist therapy can also be considered to extend the decision-making timeframe for the patient temporarily where endogenous hormone production is incongruent with gender identity.
Ethics and human rights issues
A major consideration in paediatric decision making is respecting parental views/wishes and the impact on the individual as they become more aware of their diagnosis and involved in decision making.23 Some patient groups advocate a complete moratorium on any genital/gonadal surgery until the individual is able to give informed consent.24 This questions the parents’ ability to act in the best interest of the child and whether the best interest of the child is identical to that of the future adult25 while giving due consideration to the universal human rights of non-discrimination and equality in any decision-making process. Each individual case should be discussed on its own merit, with due consideration given to available evidence on benefits versus risks for a particular procedure as well as psychosocial, cultural and personal circumstances.26 Six key ethical principles in the management of DSD were proposed by Gillam et al27 and later adopted by the consensus DSD guidelines2 to guide the decision-making process for DSD MDTs. However, striking a balance between ethical principles can be challenging as respecting one principle may result in compromise of another.
In Australia, the child is unable to consent to a surgical procedure until they reach the developmental age at which they can understand the procedure and its consequences. Until then, parents or legal guardians are responsible to give consent provided they are fully informed and make the decision in child’s best interest.28 DSD MDT meetings may enable the deferral of surgical procedures that are not deemed medically necessary until the age at which the child can consent, and may provide a viable alternative to involvement of legal agencies in the management of DSD.
Transition to adult care
People born with variations in sex characteristics often require individualised long-term hormone replacement, fertility management, sexual health counselling and ongoing psychological support. Adult MDTs for DSD are rare; therefore, handover from the paediatric MDT to specialists can be difficult to navigate. The primary care physician has a vital role, together with the paediatric team, to work through a transition checklist and link individuals with DSD to appropriate services throughout the lifespan.29 Building rapport and having sensitive discussions relating to phenotypic, anatomical and hormonal variations, gender identity and sexual health can be complex and take time.
Conclusion
Individuals with DSD may present in the newborn period with atypical genitalia, during adolescence with atypical pubertal development, or in adulthood with fertility issues. Management of DSD requires highly specialised multidisciplinary care encompassing medical, surgical, psychosocial, psychosexual and human rights/ethical aspects. Formal DSD MDT meetings provide a framework and opportunity for interactive discussion between representatives from several specialities and help create local expertise within the team. The primary care physician plays a key part in multidisciplinary care by providing holistic support to the individual and their family. Because of the rarity of DSD, national and international collaboration is necessary to carry out meaningful research. It is essential that management practices are continually evaluated and new advances are embraced as more outcome data become available from scientific research.