Adrenal diseases are generally considered to be uncommon. Indeed, ‘headline acts’ such as adrenal carcinoma, phaeochromocytoma or Cushing’s syndrome are rare; despite this, the possibility that they may be present continues to challenge clinicians. However, not all adrenal disease is uncommon: adrenal incidentalomas discovered on computed tomography (CT) scans are endemic;1 aldosterone excess contributes significantly to the burden of disease associated with hypertension;2 and the widespread use and misuse of glucocorticoids coupled with ready access to cortisol levels creates a novel set of clinical conundrums. In this commentary, the authors address the current state of the adrenal gland including the appropriate first-line investigations (Table 1).
| Table 1. Initial investigations of adrenal disease |
| Adrenal disease |
Initial investigation(s) |
| Adrenal incidentaloma |
- Dedicated adrenal CT scan to differentiate benign from malignant lesion
- To assess functionality, screen for Cushing’s syndrome, primary aldosteronism and phaeochromocytoma (see below)
|
| Cushing’s syndrome |
- 1 mg overnight dexamethasone suppression test, or
- Midnight salivary cortisol (measured twice), or
- 24-hour urinary free cortisol (measured twice)
|
| Primary aldosteronism |
- Aldosterone-to-renin ratio
|
| Phaeochromocytoma |
- Plasma metanephrines, or
- 24-hour urinary metanephrines
|
| Adrenal insufficiency |
- Early morning cortisol and ACTH
|
| ACTH, adrenocorticotropic hormone; CT, computed tomography |
Adrenal incidentaloma: A disease of imaging
A new disease, ‘adrenal incidentaloma’, arose in the 1980s, became prevalent in the 1990s and is endemic this millennium.1 It is one of many consequences of the widespread application of advanced diagnostic imaging, being a condition of ascertainment rather than disease. An adrenal incidentaloma is defined as an adrenal mass lesion >1 cm in diameter that is serendipitously discovered by radiological examination, in the absence of symptoms or clinical findings suggestive of adrenal disease.1 As for much of endocrinology, it is important to distinguish between structure (the gland) and function (hormone levels).
An incidentaloma as a structural abnormality is optimally characterised by a dedicated adrenal CT scan. For an -oma, the key question is whether the lesion is benign or malignant (Box 1). An important perspective is that while 3–10% of the population have adrenal tumours, an incidence that increases with age, adrenal carcinoma occurs in <2 cases per million per year.3 The majority of these benign lesions are lipid-rich adenomas that have a density of <10 Hounsfield units (HU) on a non-contrast CT scan. Features on a CT scan that further guide the distinction are a smooth, rounded homogenous appearance with a diameter of <4 cm. It is generally recommended that lesions >6 cm in diameter are resected, making 4–6 cm a ‘grey area’. Although the added value of a CT scan with contrast is debated, contrast media washout at 10 or 15 minutes with absolute washout >60% or relative washout >40% is consistent with a benign lesion. Magnetic resonance imaging (MRI) can be used with phase shift to demonstrate lipid content, but MRI is often less accessible and more expensive. It should be noted that most lesions >4 cm will still be benign. The European Endocrine Society guidelines recommend that if the non-contrast CT scan is consistent with a benign adrenal mass (≤10 HU, homogeneous, <4 cm), no further imaging is required.1 Where there is any ambiguity, a further CT scan in 6–12 months is suggested.
| Box 1. Causes of adrenal masses |
- Adenoma
- Carcinoma
- Phaeochromocytoma
- Congenital adrenal hyperplasia
- Massive macronodular adrenal disease
- Nodular variant of Cushing’s syndrome
- Myelolipoma
- Neuroblastoma
- Ganglioneuroma
- Haemangioma
- Lymphoma
- Metastasic malignancy
- Cyst
- Haemorrhage
- Amyloidosis
- Infiltrative and granulomatous diseases including tuberculosis
|
A benign lesion may be functionally active: the potential functional consequences of an adrenal lesion follow from the biology, with the adrenal cortex synthesising cortisol and aldosterone while the adrenal medulla produces catecholamines. Clinically this may be reflected in symptoms and signs of Cushing’s syndrome, primary aldosteronism (PA) or phaeochromocytoma respectively; however, in the context of an incidentaloma, the features are, by definition, subtle or occult. Hypertension should provoke further investigation as it is a key feature suggestive of PA or phaeochromocytoma, although hypertension may also be associated with Cushing’s syndrome. To rule out unrecognised or subclinical adrenal disease, the most pragmatic approach is to biochemically exclude each of these three possibilities for an incidentaloma. Although not specific, plasma metanephrine levels are very sensitive, as discussed later in this article in the context of hypertension. Therefore, a normal value in this context can be taken to exclude a phaeochromocytoma (which is also ruled out if the lesion is ≤10 HU).4
For PA, a serum potassium test is useful but not sensitive, so measurements of aldosterone, renin and their ratio are the optimal screening tests. Various approaches have been recommended to diagnose subclinical Cushing’s syndrome – which, despite featuring in an extensive amount of literature, is relatively uncommon. In the absence of overt Cushing’s syndrome, these authors recommend the overnight dexamethasone suppression test, in which a 1 mg dexamethasone tablet is taken at midnight followed by an 8.00 am cortisol estimation; 24-hour urinary free cortisol or midnight salivary cortisol estimation may also be used. A value of <50 nM on the overnight dexamethasone suppression test rules out Cushing’s syndrome, with values <70 nM likely to be normal. Current guidelines suggest that cortisol levels between 51 nmol/L and 138 nmol/L should be considered as evidence of ‘possible autonomous cortisol secretion’, and cortisol levels post dexamethasone of >138 nmol/L should be taken as evidence of ‘autonomous cortisol secretion’.1 It must be emphasised that this is a screening test, not a diagnostic test; it is excellent for ruling out Cushing’s syndrome but does not diagnose it. In the case of an ambiguous result, the screening test should be repeated. There are many reasons why the cortisol may apparently fail to suppress. Where suppression fails to occur, this must be further explored.
Adrenal hypertension: The underappreciated epidemic
Primary aldosteronism
PA, also known as Conn syndrome, affects 5–10% of patients with hypertension in primary care, and up to 30% of patients with resistant hypertension.5 However, PA is rarely screened for and was diagnosed in <0.1% of patients with hypertension in a survey of primary care clinics in Victoria.2 Hypokalaemia is traditionally thought of as a hallmark of the disease, but it is only present in 30% of cases, making it an unreliable clinical marker for PA.6 The patient with PA may, on presentation, be indistinguishable from a patient with essential hypertension. However, patients with PA carry a higher risk of cardiovascular complications including stroke, atrial fibrillation, coronary artery disease and heart failure when compared with patients with blood pressure–matched essential hypertension, due to the deleterious effects of aldosterone.7 It is therefore crucial to consider PA in the assessment of hypertension so that a timely diagnosis can be made to enable targeted treatment and prevention of cardiovascular consequences.
Screening for PA is performed by measuring the aldosterone-to-renin ratio (ARR) using a morning blood sample. Patients with PA have an elevated plasma aldosterone concentration and suppressed renin due to negative feedback, resulting in an elevated ARR. An ARR above the reference range (>70 pmol/L:mU/L in most laboratories) raises the possibility of PA, although the final diagnosis often requires confirmatory testing such as the saline infusion test.8 Once the diagnosis of PA is confirmed, subtyping is performed with adrenal venous sampling to differentiate bilateral and unilateral PA. Unilateral PA can potentially be cured with an adrenalectomy, while bilateral PA can be effectively treated with mineralocorticoid receptor antagonists (spironolactone or eplerenone), which specifically block the actions of excess aldosterone.
It is important to emphasise that the ARR is a screening rather than diagnostic test and can be affected by many commonly used antihpertensive medications, so patients should ideally cease medication or switched to non-interfering antihypertensives prior to testing (Table 2).9 Furthermore, confirmatory testing and subtyping usually require access to a specialised centre, and primary care physicians are encouraged to liaise with their local experts.10
| Table 2. Factors affecting the aldosterone-to-renin ratio (ARR) |
| Causes of false-positive ARR |
Beta-blocker
Central agonists (clonidine, α-methyldopa)
Nonsteroidal anti-inflammatory drugs
Licorice
Renal impairment
Oral oestrogens |
| Causes of false-negative ARR |
Diuretics
Dihydropyridine calcium channel blockers
Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers
Hypokalaemia
Dietary salt restriction
Renovascular disease
Pregnancy |
| Antihypertensive medications that are less likely to interfere with ARR |
Sustained-release verapamil
Moxonidine
Prazosin
Hydralazine |
Cushing’s syndrome
Many of the clinical manifestations of Cushing’s syndrome overlap with those of metabolic syndrome, including hypertension, obesity, diabetes, depression and menstrual irregularity. Cushing’s syndrome presenting as hypertension is usually overt, with characteristic features such as proximal myopathy, truncal obesity including intrascapular (Buffalo hump) and supraclavicular fat pads, facial plethora and violaceous abdominal striae or easy bruising.11 Screening for Cushing’s syndrome was outlined earlier in this article. These screening tests should generally be measured twice to account for the variable hypercortisolism that may be encountered in patients with mild or fluctuating disease. If any of these tests are abnormal, referral to an endocrinologist is recommended for further testing. An elevated morning serum cortisol level has limited utility in suggesting possible Cushing’s syndrome due to the diurnal nature of cortisol secretion (it is highest in the morning) and its variability within and between individuals.
Phaeochromocytoma
Phaeochromocytoma is a rare tumour with a prevalence of approximately 0.5% among patients with hypertension.12 Phaeochromocytoma refers to catecholamine-secreting tumours that arise from chromaffin cells of the adrenal medulla, while paraganglioma is the term used for a similar disease that arises from the sympathetic ganglia. Most patients with phaeochromocytoma have hypertension, while the ‘classic triad’ of episodic headache, sweating and tachycardia is only found in 25% of cases.13 It is becoming increasingly clear that a significant proportion of phaeochromocytoma are hereditary, being associated with germline mutations in the succinate dehydrogenase genes, or syndromic as part of multiple endocrine neoplasia type 2, von Hippel-Lindau syndrome or, less commonly, neurofibromatosis type 1. Asymptomatic phaeochromocytoma are being increasingly discovered through genetic testing of affected families. Measurement of plasma free metanephrines and normetanephrines using mass spectrometry–based methods has a sensitivity of 97% and a specificity of 93% for detecting phaeochromocytoma. It is the preferred screening test, although 24-hour urine fractionated metanephrines also have high sensitivity and specificity for detecting phaeochromocytoma if collected accurately.14 Minor elevations in these analytes need to be viewed with caution given that acute illness or medications such as antidepressants can lead to false-positive test results.
Following the biochemical diagnosis of phaeochromocytoma, a localisation study is usually performed with CT or MRI of the abdomen and pelvis.
15 Patients with phaeochromocytoma are initially treated with an alpha-adrenergic antagonist (eg phenoxybenzamine or prazosin). Once an adequate alpha blockade has been achieved, a beta-blocker can be added to control the tachycardia that often results from an alpha blockade. It is important that an alpha-blocker is commenced before a beta-blocker, as beta blockade alone may result in a hypertensive crisis.
Adrenal insufficiency: Real, iatrogenic and imagined
The symptoms of adrenal insufficiency are often nonspecific, including fatigue, weight loss, nausea, loss of appetite, depression and anxiety. Hence, a morning cortisol level is not uncommonly requested. A low cortisol level may be caused by primary adrenal disease or, less commonly, pituitary or hypothalamic pathology, as well as iatrogenic factors such as exogenous glucocorticoid use.
Low cortisol with high ACTH
The diagnosis of primary adrenal insufficiency (or Addison’s disease) is highly likely if an early morning (8.00–9.00 am) cortisol level is <140 nmol/L in combination with an adrenocorticotropic hormone (ACTH) concentration elevated more than two-fold above the upper limit of the reference range. Clinical features include postural hypotension and salt craving caused by mineralocorticoid (aldosterone) deficiency, in addition to hyperpigmentation of the skin and mucous membranes due to increased secretion of ACTH and melanocortin. Common biochemical features include hyponatraemia, hyperkalaemia and hypoglycaemia.16 If the morning cortisol level is >365 nmol/L, adrenal insufficiency is highly unlikely.16 For patients who do not require acute glucocorticoid treatment, the corticotropin stimulation test, or short Synacthen test, is the standard confirmatory test. Peak cortisol level below 500 nmol/L at 30 or 60 minutes after the injection of 250 μg Synacthen indicates adrenal insufficiency.17 Midnight salivary cortisol and 24-hour urinary free cortisol measurements do not aid the diagnosis of adrenal insufficiency.
The most common cause of primary adrenal insufficiency in adults is autoimmune disease, which typically occurs between 30 and 50 years of age, with autoantibodies against CYP21A2 (21-hydroxylase) being commonly detected. Other autoimmune conditions may also be present, with thyroid disease being the most common in addition to vitiligo, type 1 diabetes, primary gonadal failure, coeliac disease and pernicious anaemia.18 In individuals who are CYP21A2 autoantibody–negative, an adrenal CT scan may be useful to identify infectious or infiltrative diseases such as tuberculosis and tumours (Box 1, Figure 1).
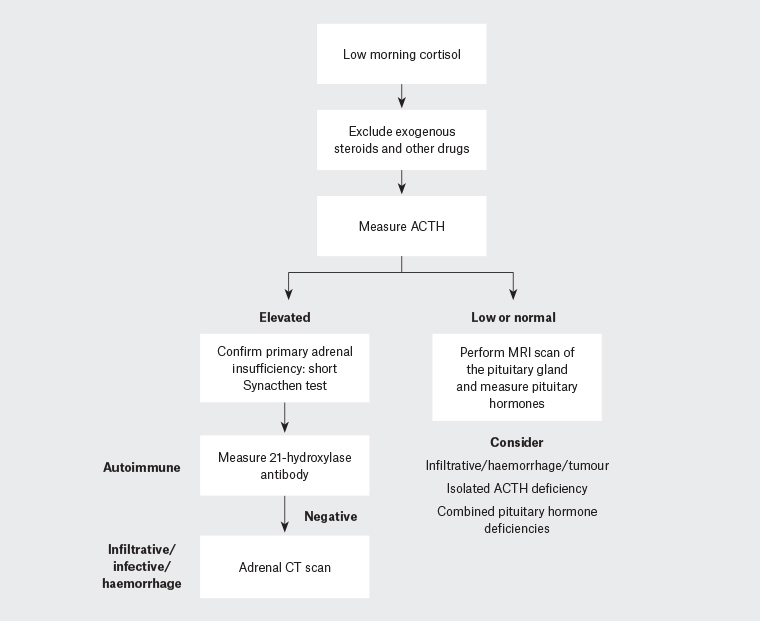
Figure 1. Pathway for the investigation of adrenal insufficiency22
ACTH, adrenocorticotropic hormone; CT, computed tomography; MRI, magnetic resonance imaging
Treatment of primary adrenal insufficiency includes both glucocorticoid replacement with hydrocortisone (15–25 mg per day) or cortisone acetate (20–35 mg per day) in two or three divided doses, and mineralocorticoid replacement with 9α-fludrocortisone (50–200 μg) once daily. As a result of adverse effects on bone and lipid profiles, prednisolone (3–5 mg once per day) is only used when compliance is poor. Modified-release glucocorticoids to replicate normal circadian rhythm are undergoing clinical trials. Dose titration is guided by postural blood pressure, body weight, energy levels and symptoms/signs of glucocorticoid insufficiency and excess. The adequacy of fludrocortisone replacement is determined by the renin level. Patient education about carrying an emergency card/bracelet and increasing glucocorticoid dosage, or seeking specialist/emergency care, in situations of stress such as gastrointestinal disturbance, infections, surgical procedures and emotional stress is crucial for the prevention of adrenal crisis.19
Low cortisol with low or suppressed ACTH
Hypothalamic or pituitary causes of adrenal insufficiency are different from primary adrenal insufficiency in that adrenal mineralocorticoid production is retained and the ACTH level is low or suppressed, so electrolyte disturbance and skin pigmentation are uncommon. Patients may display other features of hypothalamic–pituitary disease including visual field defects; measurement of anterior pituitary hormones (thyroid stimulating hormone, free thyroxine, prolactin, luteinising hormone, follicle stimulating hormone, testosterone or oestradiol, growth hormone, insulin-like growth factor 1) and a pituitary MRI will help to form a diagnosis. Glucocorticoid but not mineralocorticoid replacement will be required in addition to targeted treatment of the specific aetiology.
For patients with low cortisol and ACTH levels but no evidence of hypothalamic–pituitary disease, exogenous glucocorticoid exposure should be suspected. Synthetic glucocorticoids such as prednisolone and dexamethasone are, respectively, incompletely or not detected by cortisol assays and therefore lead to a biochemical picture of adrenal insufficiency, sometimes with incongruent clinical symptoms and signs of cortisol excess rather than deficiency. True adrenal insufficiency often follows the withdrawal of glucocorticoids and can occur with any form of administration (oral, inhalation, topical, nasal, intra-articular), dose and treatment duration.20 It is important to be aware that dexamethasone and betamethasone (long-acting) are approximately 25 times more potent than hydrocortisone (short-acting), and more likely to suppress ACTH. Sometimes meticulous history-taking is needed to identify the glucocorticoid – herbal supplements, compounded medicines, skin-whitening creams and intra-lesional injections (eg for keloid scars) can all be culprits. Patients with unexplained symptoms after steroid withdrawal should be tested for possible adrenal insufficiency with the short Synacthen test. For those with an insufficient response, treatment should be initiated with physiological doses of hydrocortisone with a plan to wean and review endogenous adrenal function over the ensuing months.
A range of other medications can lead to a low cortisol level: opioids (morphine, fentanyl, tramadol and methadone) can suppress central ACTH production; immune checkpoint inhibitors (eg ipilimumab, pembrolizumab, nivolumab) can cause hypophysitis with isolated ACTH deficiency or even panhypopituitarism; other agents can block the steroidogenic pathway (eg abiraterone for prostate cancer treatment). For patients with low cortisol due to iatrogenic causes, referral to an endocrinologist for further evaluation is advisable.
Adrenal fatigue
The term ‘adrenal fatigue’ is often used in the general media to describe an alleged condition caused by chronic exposure to stressful situations that leads to adrenal ‘overuse’ and subsequent failure. However, ‘adrenal fatigue’ is not a recognised medical condition.21 Careful evaluation of fatigue is required to identify the underlying aetiology.
Implications
Although adrenal diseases that are less common tend to attract much of the focus in this area, a radical rethink is needed. Adrenal incidentaloma can be assessed expeditiously in general practice with a dedicated non-contrast CT scan, an ARR, plasma metanephrines if indicated and an overnight 1 mg dexamethasone suppression test. This approach will identify the majority of benign lesions and avoid unnecessary specialist referral and/or investigations. These authors’ work10 and that of others has established that primary aldosteronism is a common, treatable cause of hypertension that warrants screening in all patients with hypertension,2 preferably at diagnosis. Finally, adrenal insufficiency is a life-threatening condition with non-specific symptoms that requires a low threshold of suspicion for evaluation in general practice. Novel immune-modulatory medications and obscure sources of glucocorticoids are important iatrogenic causes of adrenal insufficiency and require careful evaluation.