Erratum
Due to a production error, Cushing’s disease was incorrectly referred to as Cushing’s syndrome in the print version of the following article. The corrections have been made to the HTML and PDF versions of this article.
A full erratum was published in the March 2021 issue of AJGP. The journal apologises to the authors and our readers for these errors and any confusion they may have caused.
Pituitary tumours are surprisingly common – at least 10% of people harbour an asymptomatic lesion in their pituitary that never causes a problem.
1 Most of these lesions are small microadenomas or cystic lesions, discovered incidentally when brain imaging is done for another indication. However, there is a broad range of pathologies that may present as a sellar or parasellar mass, as outlined in Table 1.
| Table 1. Differential diagnosis of the sellar and parasellar mass27 |
| Pituitary adenomas |
- Non-functioning
- Functioning
|
| Cystic lesions |
- Rathke’s cleft cyst
- Arachnoid cyst
- Epidermoid cyst
- Dermoid cyst
|
| Non-adenomatous tumours |
- Craniopharyngioma
- Germ cell tumour
- Meningioma
- Glioma
- Pituicytoma
- Chordoma
- Metastasis
- Sarcoma
- Lymphoma
- Hypothalamic hamartoma
|
| Inflammatory/infiltrative lesions |
- Hypophysitis
- Sarcoidosis
- Langerhans cell histiocytosis
- Pituitary abscess
|
| Other |
|
Biochemical testing is used to determine function (ie to confirm or exclude both hormonal hypersecretion and hypopituitarism), while radiology (preferably magnetic resonance imaging [MRI]) is used to assess the structure, extent of mass effect and tumour invasion.
The normal anterior pituitary secretes six main hormones from five separate cell lineages:
- adrenocorticotrophic hormone (ACTH) from corticotroph cells
- thyroid stimulating hormone (TSH) from thyrotroph cells
- growth hormone (GH) from somatotroph cells
- prolactin from lactotroph cells
- follicle stimulating hormone (FSH) and luteinising hormone (LH) from gonadotroph cells.
The posterior pituitary releases two hormones, which are synthesised in the hypothalamus – arginine vasopressin (also known as antidiuretic hormone) and oxytocin.
Epidemiology
The prevalence of symptomatic pituitary tumours in the population is approximately one in 1000 people.2,3 Pituitary adenomas may be functional or non-functioning. A breakdown of pituitary adenoma subtypes is presented in Table 2. Prolactinomas are the most common pituitary tumours, comprising over 50% in some series,3 while clinically non-functioning pituitary adenomas make up approximately 30% of cases.3,4 Pituitary adenomas are defined as microadenomas if <10 mm in maximum diameter, macroadenomas if ≥10 mm in maximum diameter, and ‘giant’ pituitary adenomas if >4 cm in maximum diameter (Figure 1). Natural history studies examining the behaviour of incidentally discovered microadenomas indicate that fewer than 5% grow significantly.5 The growth rate of tumours in the first two years can be used to predict the need for later intervention.6 Incidentally discovered pituitary tumours found on imaging for other indications should be worked up as outlined in this article, looking for features of both hyper- and hypofunction, and referred to an endocrinologist for evaluation and follow‑up.
| Table 2. Pituitary adenoma subtypes27 |
| Clinically non-functioning pituitary tumours |
- Silent gonadotroph adenoma
- Other non-secretory adenomas from corticotroph, lactotroph and somatotroph cell lineage
- Null cell adenoma
|
| Functioning pituitary tumours |
- Prolactinoma
- GH-secreting adenoma (acromegaly)
- ACTH-secreting adenoma (Cushing’s disease)
- Rare functioning tumours – thyrotropinoma, FSH-secreting pituitary adenoma
|
| ACTH, adrenocorticotrophic hormone; FSH, follicle stimulating hormone; GH, growth hormone |
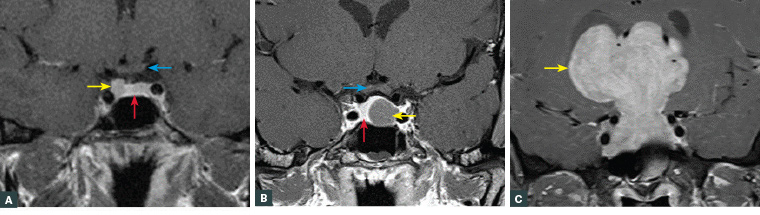
Figure 1. Magnetic resonance imaging of pituitary tumours
A. Right-sided pituitary microadenoma (yellow arrow). The normal pituitary (red arrow) and optic chiasm (blue arrow) are indicated; B. Left-sided pituitary macroadenoma (yellow arrow) with mild suprasellar extension. The normal pituitary is displaced to the right (red arrow). The optic chiasm (blue arrow) is well clear of the superior margin of the tumour; C. Giant pituitary adenoma (yellow arrow). Neither the normal pituitary nor the optic chiasm are visible.
Pituitary adenoma subtypes
Clinically non-functioning pituitary adenomas
Clinically non-functioning pituitary adenomas are characterised by an absence of any clinically significant hormonal hypersecretion and usually arise from gonadotroph cells.7 Secretion of the intact hormones (FSH or LH) is rare, but elevated alpha or beta subunits may be detected in the circulation. Tumours derived from other pituitary cell lineages can present as non-functioning adenomas, such as ‘silent’ corticotroph, ‘silent’ somatotroph or ‘silent’ lactotroph tumours.7 These tumours lack the intracellular mechanisms to actively secrete the hormone systemically. Non-functioning pituitary adenomas cause clinical problems due to mass effect (eg headaches, visual field defects) and hypopituitarism. Typically, they are not fast growing and therefore may have been present for several years before becoming apparent clinically. Treatment is surgery for those with significant mass effect or growing tumours, while small, stable lesions can be monitored without intervention.6,7 Some non-functioning tumours may behave aggressively, requiring more than one operation and/or forms of radiation therapy,7 but malignant tumours are rare.
Prolactinomas
It is important to appreciate that hyperprolactinaemia may be due to a range of other conditions such as stress, pregnancy, lactation, nipple stimulation and medications (particularly anti-emetics and antipsychotics). Up to 20% of cases may be due to ‘macroprolactin’, a biologically inactive immune complex between prolactin and immunoglobulin G.8 There is a significant sex difference between the presentations of prolactinoma. Women have a higher prevalence of microprolactinoma, which usually results in menstrual disturbance or galactorrhoea (alone or in combination). Conversely, men are more likely to have larger macroadenomas and have visual field defects and hypopituitarism at presentation.9 The explanation for the size difference is not simply due to delayed presentation in men,9 since natural history studies of microprolactinomas suggest most do not increase in size, even if untreated.10 Hyperprolactinaemia is discovered in approximately 15% of cases of secondary amenorrhea in women of reproductive age,11 and the measurement of serum prolactin should be undertaken in all women presenting with menstrual disturbance or infertility. The treatment of choice for the majority of patients with prolactinoma is a dopamine agonist, most commonly cabergoline. This normalises hyperprolactinaemia in 85% of cases and results in shrinkage in 80% of cases.8 In experienced hands, surgery can be considered for primary therapy for patients with microadenomas and for women with macroadenomas contemplating pregnancy,12 but it is usually reserved for patients either resistant to or experiencing significant side effects from the dopamine agonist.8
Acromegaly
Acromegaly is the clinical syndrome caused by excessive GH secretion. If GH excess occurs prepubertally, gigantism arises, characterised by excessive growth.13 The key issue for general practitioners (GPs) is recognition; the clinical features of acromegaly are listed in Table 3. For a patient with suggestive clinical features, the screening test is measurement of serum insulin-like growth factor-1 (IGF1).14 Circulating IGF1 arises predominantly from the liver and reflects integrated GH secretion. In contrast, measuring a random GH alone is not helpful as, because of its pulsatile nature, ‘high’ levels may indicate a secretory pulse in an unaffected individual. A significantly elevated IGF1 (>1.1 times the upper limit of the age-matched reference range) should then be a trigger for referral to an endocrinologist for further investigation and management. The treatment of choice is usually surgery, but since the majority of cases are caused by macroadenomas (often invasive), multimodality treatment – including radiation therapy and medical therapy – is frequently required.13 Long-acting injectable somatostatin analogues are the most frequently used medications. They act directly on the tumour, reducing GH secretion and often inducing tumour shrinkage. An alternative is pegvisomant, a GH receptor antagonist administered by a daily injection that blocks the effects of GH but does not reduce GH secretion.13 Young people aged under 30 years with acromegaly or gigantism should undergo genetic testing.15 Patients with acromegaly are best managed in the setting of a well-established multidisciplinary team.16
| Table 3. Clinical features of hormonal excess in functioning pituitary adenomas27 |
| Condition |
Clinical features |
| Prolactinoma |
- Galactorrhoea
- Menstrual disturbance (oligomenorrhoea/amenorrhoea)
- Male hypogonadism (low libido, erectile dysfunction)
|
| Acromegaly |
- Enlargement of hands and feet
- Frontal bossing
- Splayed dentition
- Mandibular enlargement – jaw malocclusion
- Enlarged tongue
- Skin tags
- Oily skin
- Sweating
- Hirsutism (women)
- Obstructive sleep apnoea
- Osteoarthritis
- Carpal tunnel syndrome
- Hypertension
- Cardiomyopathy
- Goitre
- Liver/spleen enlargement
- Colonic polyps
- Diabetes
- Tall stature (prepubertal onset = gigantism)
|
| Cushing’s disease |
- Thin skin
- Easy bruising
- Proximal myopathy
- Broad (>1 cm) purple striae (abdomen, flanks, breasts, thighs, arms)
- Osteoporosis/fracture (particularly vertebral fracture)
- Central adiposity
- Dorsocervical (‘buffalo hump’) and supraclavicular fat pads
- Facial plethora
- ‘Moon’ facies
- Hirsutism (women)
- Menstrual disturbance
- Psychiatric/mood disorders
- Hypertension
- Diabetes
- Pityriasis versicolor
- Short stature/growth failure (children)
|
Cushing’s disease
The most common cause of a Cushingoid appearance is secondary to exogenous administration of glucocorticoids. Approximately 70% of cases of endogenous Cushing’s syndrome are caused by an ACTH-secreting pituitary adenoma (known as Cushing’s disease).17 Again, the key issue here for GPs is recognition and screening. The predominant clinical features of Cushing’s disease are listed in Table 3, with the first five being more specific. Patients with simple obesity, hypertension and type 2 diabetes should not be routinely screened for Cushing’s disease unless they display some of the specific features. Measures of morning cortisol and ACTH are not useful for diagnosing Cushing’s syndrome, but once Cushing’s syndrome is confirmed, measurement of ACTH is essential to guide further investigations because a suppressed ACTH indicates an autonomous adrenal source. While the Endocrine Society Guidelines suggest three possible screening tests,18 for most patients in the primary care setting, the 1 mg overnight dexamethasone suppression test (1 mg DST) is the best screening test. A post-dexamethasone morning cortisol of <50 nmol/L essentially excludes Cushing’s syndrome, and rarely is further testing required. However, a positive test does not diagnose Cushing’s syndrome, and referral to an endocrinologist is recommended for evaluation that is more detailed. Instances in which the 1 mg DST is not suitable include when a patient is taking CYP3A4 enzyme–inducing medications such as carbamazepine (increased metabolism of dexamethasone, leading to non-suppression of cortisol) or they are taking oral oestrogens, including the oral contraceptive pill (increased serum cortisol from oestrogen stimulation of corticosteroid-binding globulin).19 In these cases, 24-hour urinary free cortisol or late night salivary cortisol are preferred, but at least two samples are required. Cushing’s syndrome may be caused by pituitary, adrenal and ectopic sources. The key steps are: 1) establishing that the patient has a pituitary source, 2) determining if a post-surgical remission has been achieved and 3) managing patients for whom surgery does not result in remission. Recurrence rates are high, even after successful surgery (approximately 25% overall); therefore, lifelong follow-up is required.20 Management of Cushing’s disease is optimal in a specialised multidisciplinary pituitary service.
Rare functioning pituitary adenomas
Rarely, pituitary tumours may secrete TSH or FSH. Patients with thyrotropinomas present with hyperthyroidism with a normal or elevated TSH in the presence of an elevated free thyroxine level, in contrast to autonomous thyroid function.21 FSH-secreting adenomas are very rare and present in women of pre-menopausal age with ovarian hyperstimulation, sometimes with marked ovarian and uterine enlargement, and dysfunctional uterine bleeding.22
Non-adenomatous pituitary and parasellar masses
As outlined in Table 1, there is a wide range of causes of a sellar or parasellar mass. These may present with symptoms of mass effect such as visual loss or pituitary dysfunction, or be discovered incidentally on imaging for non-related reasons. The Rathke’s cleft cyst, resulting from failure of the obliteration of Rathke’s pouch during embryological development, is one of the common non-adenomatous lesions.23 Small Rathke’s cleft cysts may be managed conservatively, with surgery reserved for those causing symptomatic mass effect or pituitary dysfunction.23 A detailed discussion of the other lesions is beyond the scope of this article.
Red flags that indicate that a lesion is unlikely to be a pituitary adenoma include:
- diabetes insipidus (thirst and polyuria, with inability to concentrate the urine)
- upper cranial nerve palsies III, IV and VI, causing ptosis/eye movement disorders. Generally this will only occur in patients with pituitary adenomas who have undergone pituitary apoplexy.
Hypopituitarism
Pituitary hormonal dysfunction is unusual in microadenomas but more common as tumour size increases. The usual mechanism is obstruction (by the tumour itself or increased intrasellar pressure) of hypothalamic releasing factors. These factors are transported via the hypothalamic–hypophyseal portal circulation to the normal anterior pituitary gland.24 Therefore, many patients have the potential to regain hormone function following tumour resection by a skilled pituitary surgeon. Sometimes treatment of a pituitary or parasellar tumour with surgery or radiotherapy may result in loss of pituitary function due to direct damage to the normal anterior pituitary/hypothalamus. Each hormonal axis must be assessed individually and replaced if found to be deficient.
A standard pituitary function panel drawn in the morning before 9.00 am is outlined in Box 1.
| Box 1. Pituitary function test panel |
- Electrolytes/urea/creatinine/glucose
- Cortisol (± ACTH)
- Free thyroxine, TSH
- Prolactin
- FSH/LH
- Oestradiol (women), testosterone (men)
- Insulin-like growth factor 1 (± GH)
|
| ACTH, adrenocorticotrophic hormone; FSH, follicle stimulating hormone; GH, growth hormone; LH, luteinising hormone; TSH, thyroid stimulating hormone |
Results that may point to the presence of a pituitary lesion, whereby the absence of a pituitary hormone response to low target organ hormone levels indicates secondary (pituitary/hypothalamic) hypofunction, include:
- low thyroxine with inappropriately low or normal TSH, which indicates secondary hypothyroidism, in contrast to the elevated TSH seen in primary hypothyroidism
- low testosterone or oestrogen with inappropriately low or normal FSH/LH, or pre-menopausal FSH/LH levels in a woman post-menopause. This is secondary hypogonadism, and while functional causes exist with no structural pituitary abnormality, it still requires further investigation.
- isolated mild-to-moderate hyponatraemia – commonly seen in secondary adrenal insufficiency. It is important to measure morning cortisol levels in patients with hyponatraemia.
Assessing for secondary adrenal insufficiency and GH deficiency is more involved than assessing the other axes, often requiring stimulation tests. Reference ranges and diagnostic cut-offs for cortisol in particular vary considerably according to the assay used. The short Synacthen test is available through private pathology laboratories. While it is not 100% accurate for diagnosing secondary adrenal insufficiency, particularly if it has a recent onset, it is the most commonly used test of pituitary–adrenal function.24,25 GH is now reimbursed by the Pharmaceutical Benefits Scheme for adults with GH deficiency, but a stimulation test is required to confirm the diagnosis; this should be performed in a specialised endocrine testing centre.
For a comprehensive discussion of replacement therapy in hypopituitarism, refer to Fleseriu et al.25
Pituitary apoplexy
Pituitary apoplexy is a genuine endocrine emergency that may occur in up to 10% of patients with pituitary adenomas. It usually presents with a sudden onset of headache and hypopituitarism, with or without visual loss, eye movement disorders and an altered level of consciousness.26 The cause is acute pituitary haemorrhage or infarction. Many of the patients with pituitary apoplexy are not previously known to have a pituitary adenoma, and the diagnosis is often made when a computed tomography scan is performed to exclude a subarachnoid haemorrhage. These patients require urgent glucocorticoid replacement and should be jointly managed by endocrinology and neurosurgery, with surgery indicated if the visual changes do not improve with conservative management.26
Key points
- Pituitary conditions cover a broad spectrum of aetiologies.
- It is critical that GPs have an appropriate index of suspicion and are familiar with the basic hormone screen for hypopituitarism.
- Screening tests for causes of pituitary hyperfunction include prolactin for galactorrhoea/amenorrhoea in women or low testosterone with low or normal FSH/LH in men, IGF1 for acromegaly and 1 mg DST for Cushing’s syndrome.
- Referral to an experienced pituitary centre for multidisciplinary management is recommended.