Multiple sclerosis (MS) is a multifocal central nervous system (CNS) disorder characterised by inflammatory demyelinating lesions affecting white and grey matter. Steady accumulation of damage over time leads to irreversible disability characterising the advanced stages of the disease. The range and effectiveness of disease-modifying therapies (DMTs) have greatly expanded over the past decades, improving prognosis for patients with MS. This article covers the topics of diagnosis, DMT and prognosis of MS.
Investigations and diagnosis
MS is diagnosed by a combination of clinical and radiological features. The 2017 McDonald criteria are the standard used for diagnosis of all forms of MS.1 There are five key principles underlying diagnosis:
- A syndrome ‘typical’ for MS (refer to Table 1)
- Objective evidence of CNS involvement (almost always magnetic resonance imaging [MRI] of the brain and spinal cord confirming demyelinating lesions)
- Dissemination in space – lesions affecting multiple areas of the CNS
- Dissemination in time – multiple clinical demyelination events, or MRI showing both contrast-enhancing lesions (indicating acute demyelination) and non–contrast enhancing (indicating chronicity)
- No better explanation for the symptoms.
| Table 1. Common sites, signs and symptoms of clinically isolated syndrome or multiple sclerosis relapses1 |
| Site |
Condition |
Symptoms |
Signs |
| Optic nerve |
Optic neuritis |
Pain on eye movement, blurred vision |
Reduced monocular visual acuity, colour desaturation
Fundoscopy may be normal or may show a swollen optic disc |
| Cerebrum |
Focal supratentorial syndrome |
Dependent on cerebral location (eg hemianopia in an appropriately located occipital lesion) |
| Cerebellum |
Cerebellar disease |
Unsteadiness |
Limb or gait ataxia; horizontal or torsional gaze-evoked nystagmus |
| Spinal cord (usually multifocal and asymmetric) |
Partial myelitis affecting pyramidal tracts |
Upper or lower limb weakness
|
Pyramidal distribution weakness |
| Partial myelitis affecting spinothalamic, posterior columns |
Unilateral or bilateral limb numbness or paraesthesias; Lhermitte’s phenomenon |
Sensory level
|
| Any spinal cord lesion |
Urinary frequency or urge incontinence, constipation erectile dysfunction |
|
| Brainstem – medial longitudinal fasciculus |
Internuclear ophthalmoplegia |
Blurred or double vision |
Internuclear ophthalmoplegia |
| Brainstem – pyramidal tracts, spinothalamic and posterior columns |
Similar to that described in spinal cord |
MRI of the brain and spinal cord with post-gadolinium sequences is the investigation of choice for suspected MS or clinically isolated syndrome (CIS). As a result of Medicare Benefits Schedule restrictions, the MRI will generally be requested by a neurologist. Advances in MRI scanners and post-acquisition image analysis have led to improved diagnostic accuracy.2 Figure 1 shows examples of typical MS changes on MRI.
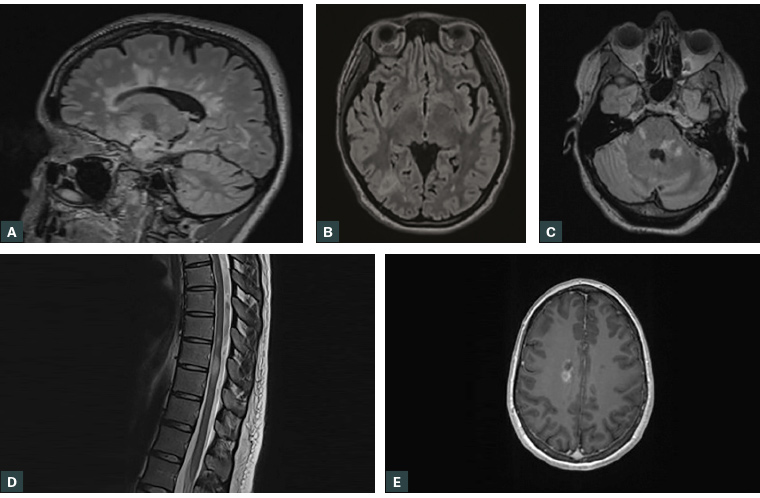
Figure 1. Typical multiple sclerosis magnetic resonance imaging findings, sourced from multiple patients
A. Periventricular lesions (FLAIR sequence); B. Juxtacortical lesion in the right parieto-occipital region (FLAIR sequence); C. Brainstem and cerebellar lesions (FLAIR sequence); D. Spinal cord lesions in the thoracic cord (T2-weighted turbo spin echo sequence); E. Gadolinium-enhancing lesion (T1-weighted sequence)
FLAIR, fluid attenuated inversion recovery
Cerebrospinal fluid (CSF) examination may show restricted oligoclonal bands – immunoglobulins present in the CSF but not the serum, indicating antibody production in the CNS. They can be a useful diagnostic aid in atypical or early cases. However, they are not a diagnostic requirement, nor are positive bands specific for MS. Research is underway for specific CSF and blood biomarkers, which may help predict future disease activity.3
Differential diagnosis
The varied presentation of MS leads to a broad range of possible differentials depending on the CNS location involved. Misdiagnosis of MS commonly occurs when presentations atypical for MS are linked with non-specific white matter lesions on MRI.4 White matter lesions are common, occurring in a quarter of people aged <45 years,5 and should be interpreted with caution on the basis of the cause for initial investigation. Red flags for an alternative diagnosis include systemic and constitutional symptoms, neurological signs outside the CNS (particularly peripheral neuropathy) and atypical MRI findings.6
One multicentre study found that the most common entities mistaken for MS were migraine (22%), fibromyalgia (15%), nonspecific symptoms with abnormal MRI (12%), functional neurological disorder (11%), and neuromyelitis optica (NMO; 7%).4
Non-MS demyelinating diseases include NMO spectrum diseases, anti–myelin oligodendrocyte glycoprotein disease (a recently described condition similar to NMO) and acute disseminated encephalomyelitis. Diagnosis is based on specialist assessment of clinical, radiological and laboratory features, and treatment with conventional MS therapies is ineffective.
Natural history and disease phases
Clinically isolated syndrome
CIS refers to a solitary episode of neurological disturbance due to CNS demyelination in the absence of fever, infection or encephalopathy.7 The disturbance is monophasic, reaching a peak within days to a few weeks and lasting at least 24 hours. Typical presentations include monocular optic neuritis, focal supratentorial syndrome, brainstem or cerebellar syndrome, or partial myelitis (Table 1). Careful history-taking for previous events is required to avoid possibly misclassifying relapsing MS.
With the advent of highly effective DMTs, there is a greater emphasis on earlier identification of MS to limit disability, making CIS a dwindling concept. Changes to the 2017 modified McDonald diagnostic criteria for MS result in some patients with a first clinical demyelinating event being diagnosed with MS if certain criteria are simultaneously met.1
MRI is the best imaging modality to prognosticate progression from CIS to MS. Over 20 years of follow up, the risk of a second attack is 80% if MRI demonstrates typical lesions and 20% if it does not.7 The risk of early MS (diagnosis within four years) increases with lesion load, especially with infratentorial lesions or more than three periventricular lesions. Overall, approximately one-third of patients have no further demyelinating events even after 30 years of follow up.
Radiologically isolated syndrome
MRI of the brain or spine performed for another reason may incidentally find demyelinating lesions. Where these lesions are in a typical distribution for MS, and detailed history and examination does not find an episode of CIS, then the presentation is termed radiologically isolated syndrome (RIS).8 Other conditions may cause similar MRI changes, particularly migraine and small vessel disease, so diagnosis requires neurologist review.
After 10 years of follow up, the probability of a patient with RIS developing a first clinical event is 51%.9 Risk factors for conversion to relapsing clinical disease are similar to that for CIS.
Relapsing remitting MS
Relapsing remitting MS (RRMS) is defined by episodes of CNS demyelination separated by time and by space (ie lesions in more than one region of the CNS). In untreated patients, ongoing neuro-inflammation results in accelerated brain atrophy of 0.5–1.4% per year,10 and relapses occur erratically, with a mean rate of 0.65 clinical attacks per year.11 Subclinical disease activity, which is the development of new lesions on imaging without clinical correlate, occurs more commonly.
An MS relapse is defined as symptoms or objective signs typical of an acute inflammatory CNS demyelinating event lasting at least 24 hours. They are caused by either a new focal lesion or reactivation of an old lesion.12 Complete symptomatic resolution from a relapse often occurs; however, mild residual signs may persist in some patients. Hyperacute onset over seconds to minutes is unusual and should prompt investigation for other causes.
Pseudorelapse occurs when other medical conditions, particularly infections, cause transient neurological worsening related to historical demyelinating damage. Relapse may be distinguished from pseudorelapse by a lack of a trigger and by symptoms localising to a previously unaffected region in the CNS.
Progressive MS
Progressive MS describes an insidious deterioration of neurological function associated with new symptoms and signs, progressing for at least one year. It is predominantly driven by neurodegeneration, with widespread neuroaxonal loss in the white and grey matter.7,13 Patients do not progress in a uniform manner, and the disease can remain relatively stable over periods of time.13 Diagnosis is made retrospectively.
Secondary progressive MS (SPMS) describes a previously relapsing remitting disease course transitioning to progressive disability. The underlying mechanism that drives transition from RRMS to SPMS is not well established.13 DMT helps to slow or prevent this transition. There is often a period of uncertainty regarding when patients transition from RRMS to SPMS.13
A small proportion of patients with MS have a progressive course from disease onset; this is termed primary progressive MS (PPMS). The most common presentation is with a gradual onset spastic paraparesis, followed by cerebellar or hemiplegic symptoms. It remains unclear whether PPMS is a separate entity from SPMS, or if it follows an unrecognised or subclinical inflammatory phase.7,13
Although patients are classified as having either relapsing or progressive MS, the driving pathology is thought of as a spectrum. Autopsies of patients with RRMS have shown widespread grey matter atrophy from early in the disease course,13 and patients with progressive disease may continue to have new enhancing lesions on MRI (called active progressive MS).
Treatment
Acute relapses
Corticosteroids are the mainstay treatment for acute relapses. Intravenous methylprednisolone is generally used, although oral methylprednisolone has been shown to have similar efficacy.14 Other options include adrenocorticotropic hormone or plasma exchange. Medical treatment is only necessary if the relapse symptoms have a significant impact on quality of life, as treatment may hasten the recovery from a relapse but not the likelihood of recovery or permanent disability.14
In cases of pseudorelapse, treatment should be targeted towards the exacerbating disease, with no role for steroids.
Disease-modifying therapies
The range of DMTs for MS continues to grow. The agent chosen is based on a combination of patient factors (age, comorbidities, plans for pregnancy), disease factors (number and location of lesions) and patient preferences (medication side effects versus efficacy). In all cases, DMTs should be initiated or modified under neurologist guidance. One DMT is used at a time – there have been no proven benefits to combination therapy. The Pharmaceutical Benefits Scheme (PBS) only covers DMTs for RRMS and a single agent for SPMS, with no agents covered under the scheme for CIS, RIS or PPMS. DMTs are assessed in trials for their effect on annualised relapse rate (ARR), disability worsening, radiological lesion load and brain volume measurements. A summary of currently available DMTs is provided in Table 2.
| Table 2. Disease-modifying therapies for multiple sclerosis14 |
| Medication |
Mode of action |
Route and recommended dose |
Monitoring |
Notable adverse effects |
Other comments |
| Interferon beta-1a |
Immunoregulatory actions including antagonism of interferon gamma, reduction of cytokine release and augmentation of suppressor T cell function |
Avonex: IMI, 30 µg weekly
Rebif: SC, 44 µg three times a week |
FBE and LFTs at one, three and six months after initiation, then annually
|
Injection-site reactions, influenza-like illness |
Dose titration recommended
Paracetamol may reduce influenza-like symptoms
Presence of neutralising antibodies may reduce response |
| Peginterferon beta-1a |
SC, 125 µg fortnightly |
| Interferon beta-1b |
SC, 62.5–250 µg every two days |
| Glatiramer acetate |
Synthetic polypeptide; possibly blocks presentation of myelin antigens to T lymphocytes |
SC, 40 mg three times a week |
None |
Injection-site reactions, post-injection systemic reactions |
Antibodies to glatiramer develop in all patients and are not associated with adverse effects or decreased efficacy |
| Teriflunomide |
Prodrug of leflunomide; inhibits pyrimidine synthesis in leucocytes |
Orally, 14 mg daily |
FBE and LFTs, monthly for six months, then every 8–4 weeks
Blood pressure regularly |
Nausea, diarrhoea |
Teratogenic – highly effective contraception must be in place
In case of toxicity or pregnancy, washout procedure with colestyramine can be used |
| Dimethyl fumarate |
Antioxidative, immunomodulatory and anti-inflammatory effects via activation of the nuclear factor (erythroid-derived 2)-like 2 transcriptional pathway |
Orally, 120 mg daily then weekly uptitration to maintenance dose of 240 mg bd |
FBE every 3–6 months, LFTs every 6–12 months
Urinalysis annually |
Flushing, nausea, vomiting, diarrhoea, abdominal pain |
Gastrointestinal upset and flushing are common and tend to improve with time. |
| Fingolimod |
Sphingosine 1-phosphate receptor modulators; prevent lymphocyte trafficking through the lymph node and cause a reversible lymphopaenia |
Orally, 0.5 mg daily |
Lymphocyte count, LFTs, blood pressure periodically
Visual acuity periodically
Skin cancer screen annually |
First-dose bradycardia, macular oedema |
In-hospital cardiac monitoring required for first dose |
| Siponimod |
Orally, 2 mg daily after initial uptitration |
Similar to fingolimod |
Cardiac monitoring in patients at risk for first dose |
| Ozanimod |
Orally, 0.92 mg daily after initial uptitration |
Cardiac monitoring in patients at risk for first dose |
| Cladribine |
Synthetic adenosine analogue, interferes with DNA synthesis, reduces circulating lymphocytes |
Two courses 12 months apart – total oral dose 1.75 mg/kg per course, half over five days in week 1, remainder in week 5 |
Lymphocyte counts at two and six months after starting treatment |
Lymphopaenia, leukopenia, neutropenia, infection, hypersensitivity, headache, rash, alopecia |
If lymphocytes
<500 cells/mm3, monitor closely for infection; if
<200 cells/mm3, consider herpes virus prophylaxis |
| Natalizumab |
Recombinant humanised monoclonal antibody to α4-integrins, inhibiting leucocyte migration from blood to CNS |
IV, 300 mg over one hour, given every four weeks (some circumstances can be given every six weeks) |
MRI brain every 6–12 months
JCV antibody testing every six months |
PML |
Persistent neutralising antibodies associated with reduced efficacy and increased risk of allergic reactions |
| Ocrelizumab |
Humanised monoclonal antibody to CD20, depletes B lymphocytes |
IV, 600 mg over several hours, every six months |
None |
Hypersensitivity and infusion reactions, infection, neutropenia
Rare: PML |
Long-term risks are not fully known, but there is a long-term risk of hypogammaglobulinaemia |
| Ofatumumab |
Fully human monoclonal antibody to CD20 |
SC, 20 mg monthly |
None |
Hypersensitivity and infusion reactions, infection, neutropenia |
Long-term risks are not fully known |
| Alemtuzumab |
Humanised monoclonal antibody to CD52 (present on B and T lymphocytes) |
Two courses 12 months apart; IV infusion over four hours, 12 mg daily for five days on the first course and three days on the second |
Enrolment in a monitoring program with monthly blood tests and urinalysis
Annual skin cancer screening |
Infusion-related reactions, autoimmune disorders |
Autoimmune disorders may be induced – thyroid dysfunction (30–40%), immune thrombocytopenic purpura, cytopenias, nephropathies, autoimmune hepatitis
Herpes virus prophylaxis recommended from the start of each treatment for 1–2 months |
| bd, twice per day; CNS, central nervous system; FBE, full blood examination; IMI, intramuscular injection; IV, intravenous; JCV, John Cunningham virus; LFTs, liver function tests; MRI, magnetic resonance imaging; PBS, pharmaceutical benefits scheme; PML, progressive multifocal leucoencephalopathy; SC, subcutaneous injection |
There are two general approaches to initiating DMTs. In the step-up approach, a modestly effective initial agent is commenced; if disease progression occurs, then a more efficacious medication is substituted. A step-down approach involves starting with a high-efficacy treatment, which may be stepped back following a period of disease stability. Which strategy more effectively balances the risk of side effects with that of reduced future disability is the subject of active research.15
Subcutaneous and intramuscular DMTs
Interferon beta and glatiramer acetate are the longest established medications used for treatment of RRMS, giving them the greatest depth of safety data.16 ARR is approximately 30% reduced when compared with placebo for each agent.17,18
Oral DMTs
Teriflunomide reduced ARR by 31% when compared with placebo, and significantly affected several MRI parameters.19 It is teratogenic, so reliable contraception is required for women of reproductive age.
Dimethyl fumarate reduced ARR by 44% when compared with placebo.20 Common adverse effects are flushing and gastrointestinal upset, which can be limited by aspirin use and taking medications with full meals.
Fingolimod, siponimod and ozanimod are oral sphingosine-1-phosphate receptor modulators. In their pivotal trials, fingolimod reduced ARR by 54% when compared with placebo,21 and ozanimod reduced ARR by 49% when compared with interferon beta-1a.22 Siponimod modestly slowed disability accumulation in patients with SPMS, especially those with relapses within two years of beginning treatment.23 First-dose electrocardiography monitoring in hospital is required for all patients starting fingolimod because of the risk of symptomatic bradycardia, but it is only required for those with pre-existing cardiac conditions for siponimod and ozanimod. Ophthalmology review prior to and 3–4 months after the commencement of fingolimod is required to exclude macular oedema.
Cladribine is given as two courses of treatment with a one-year gap between courses, with no planned ongoing treatment following this. ARR is reduced by 57% when compared with placebo.24
Intravenous DMTs
Natalizumab reduced the rate of disability progression, the ARR and the number of new gadolinium-enhancing lesions by 54%, 68% and 92%, respectively, when compared with placebo.25 The most serious adverse effect is progressive multifocal leukoencephalopathy, a life-threatening opportunistic brain infection caused by the John Cunningham virus (JCV). This risk is less than 0.07 per 1000 patients if JCV antibodies are negative, rising to 1.7–2.7% over six years with positive antibodies.26 For this reason, antibody levels are tested every 6–12 months.
Ocrelizumab is given every six months and reduced ARR by 46% when compared with interferon beta-1a.27 In addition, it is the only DMT to have shown positive effects in a patient cohort with PPMS, with minor improvements in walking speed, brain lesions and brain volume loss.28 Ofatumumab has a similar mechanism of action and efficacy as ocrelizumab but is delivered by a subcutaneous route monthly, avoiding hospital admissions for infusions.29
Alemtuzumab is given as two cycles of infusions one year apart. It reduced ARR by 55% when compared with interferon beta-1a.30 However, it may induce significant autoimmune adverse effects including idiopathic thrombocytopenic purpura, nephropathy and thyroid dysfunction. Strict laboratory monitoring is required for five years after treatment.
Emerging options and future directions
Autologous haematopoietic stem cell transplant shows great promise for the management of treatment-refractory disease.31 Treatment-related toxicity, including possible autoimmunity and infertility, is a limiting factor, so patient selection is very important. Other areas of investigation include remyelination therapies to repair damaged neurons and various neuroprotective methods to prevent degeneration of previously demyelinated nerves.
Supplements and dietary modification
Low serum vitamin D levels are associated with higher disability in patients with MS.32 Serum vitamin D should be checked regularly, with patients starting oral supplementation when levels drop below 100 nmol/L.
Altered gut bacteria and their short-chain fatty acid production have been suggested as part of MS pathogenesis. Early studies on short-chain fatty acid supplementation are promising,33 but more data are required. The Mediterranean diet has shown a non-significant trend towards improved outcomes.34 Other diets have been assessed in multiple studies with conflicting results.32 Biotin supplementation showed early promise to slow disability in progressive MS, but a recent phase III trial failed to show any benefit.35
Monitoring of treatment efficacy and treatment complications
Routine monitoring with MRI for subclinical radiological activity is often performed annually, and it may be performed more frequently when a risk of serious treatment-related adverse effects exists.2 Patients should also have an MRI scan after a suspected MS relapse. Ideally, imaging comparisons are made on scans performed using comparable MRI field strength and imaging protocols, as variations may limit image interpretation or require a new baseline scan. Spinal cord MRI is not as sensitive to changes as MRI of the brain and is not always performed after the baseline scan. Gadolinium contrast allows increased detection of active demyelination and is given when relapse or treatment failure is suspected. Each DMT has different additional monitoring requirements, listed in Table 2.
Pregnancy and breastfeeding considerations
The risk of MS relapse is reduced during pregnancy when compared with the pre-pregnancy period;36 however, the risk increases to greater than baseline in the first six months postpartum. Breastfeeding may slightly reduce this rate.36 The most important factor affecting relapses during pregnancy is pre-pregnancy disease activity.
Any patient considering pregnancy should receive expert preconception planning regarding management of their DMT through the pregnancy and postpartum to balance DMT risks with that of disease recurrence. Expert opinion may allow the use of particular DMTs during some stages of pregnancy, or with a gap between DMT use and conception.37
COVID-19 and other vaccines
As a result of their immunosuppressed state, patients with MS on certain DMTs may be at higher risk of complications from COVID-19. Vaccination against COVID-19 is recommended by both international and Australian MS medical societies for all patients with MS; however, the patient’s DMT may influence the recommended timing of vaccination, and specialist advice from the treating neurologist should be sought.
Similarly, all patients with MS are recommended to complete the routine vaccination schedule. An exception is live vaccines, as these may present a particular risk, and expert opinion should be sought prior to administration. As advice in this area is rapidly evolving, it is recommended that readers follow updated advice on this topic on the MS Australia website (www.msaustralia.org.au/covid-19-vaccination-guidance-for-people-with-ms-2).
Prognosis of established MS
The outlook for patients with MS has improved substantially over the past few decades with improved DMTs. It is difficult to accurately forecast expected outcomes for patients on DMTs, as the long-term effects of these agents on prognosis and disease progression are only now being seen. A recent review over a 16-year period showed 18% of patients with RRMS progressed to SPMS, and 11% acquired sufficient disability to require a walking aid.38 Increased morbidity is present, with an estimated 13.1 quality-adjusted life years lost per patient.39 Modifiable risk factors that have a proven association with disease progression are low vitamin D level and current smoking.32 Table 3 lists non-modifiable factors associated with RRMS prognosis.
The progressive forms of MS have significantly earlier onset of irreversible disability, and most patients have mild-to-moderate disability by the time of diagnosis.11 Risk factors associated with progression from RRMS to SPMS include older age at MS onset, male sex, high relapse frequency early in the disease, longer disease duration, higher baseline disability score, higher lesion burden on imaging, spinal cord involvement and lower brain volume.13 Despite the advancements in RRMS treatment, options for progressive disease remain limited.
The largest international registry (MSBase) to track MS prognosis is ongoing and is being performed by Australian clinicians.40
| Table 3. Early clinical and imaging features affecting prognosis in multiple sclerosis1,6 |
| Better prognosis |
Poor prognosis |
- Optic neuritis or isolated sensory symptoms as the initial presentation
- Complete recovery from the first neurological episode
- Long interval to second relapse
- No disability after five years
- Normal initial MRI
- Older age at disease onset
|
- High relapse rate in first 2–5 years
- Substantial disability after five years
- Abnormal initial MRI with large lesion load
- Infratentorial or spinal lesions on MRI
|
| MRI, magnetic resonance imaging |
Conclusion
MS is a clinical and radiological diagnosis based on the 2017 McDonald criteria. The range and effectiveness of treatment options have greatly improved over the past several decades, which has significantly improved the prognosis of MS in the modern era.
Resources