Joint hypermobility describes physiological joint movement in excess of the mean range considering a person’s age, gender and ethnicity.1 Hypermobility can be found in one to many joints and can affect peripheral joints only or affect many joints of the appendicular and axial skeleton. In the latter case, it is termed generalised joint hypermobility (GJH). Assuming that 95% of the population have normal joint mobility, those in the top 5% of the spectrum will include both asymptomatic individuals with inherited or acquired hypermobility (eg gymnasts and dancers) and individuals whose hypermobility is a manifestation of a heritable disorder of connective tissue.2 Figure 1 describes the assessment of GJH using the Beighton score and the cut-off scores for children and adolescents.
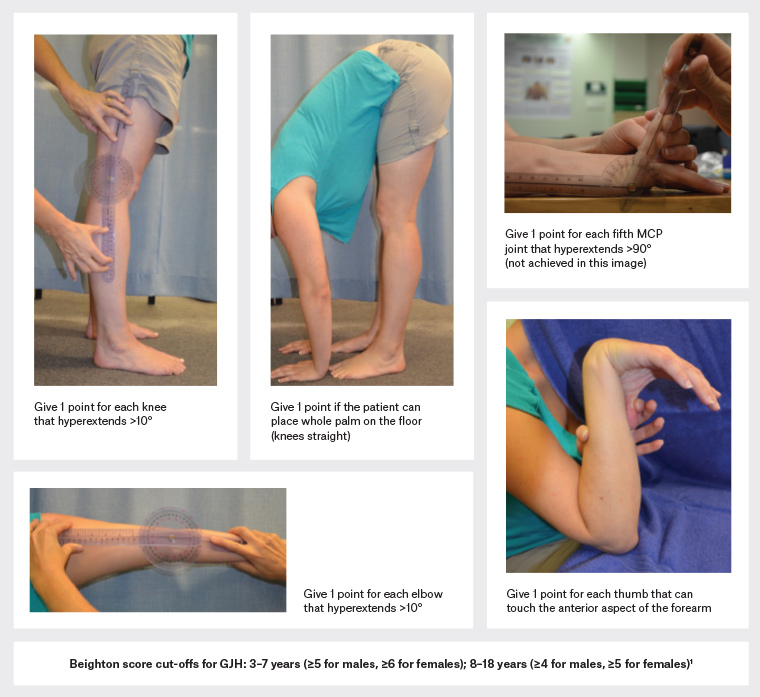
Figure 1. The five tests that comprise the Beighton score, and cut-off scores for determining generalised joint hypermobility (GJH).1 If the joint easily meets the relevant criterion on visual inspection (eg the right elbow hyperextends well past 10°), there is no need to use a goniometer. One point is allocated if the joint exceeds the criterion noted in the figure. Adding the scores from both knees, elbows, fifth metacarpophalangeal (MCP) joints, thumbs and the palms to floor tests, the maximum score is 9. Click here to enlarge.
What are hypermobility syndromes?
Hypermobility syndrome refers to a combination of signs and symptoms incorporating multiple body systems, including the musculoskeletal, cardiovascular, gastrointestinal, urogenital and neurological/autonomic systems, where joint hypermobility is a key feature.
Hypermobility syndromes are manifestations of hereditary disorders of connective tissue (HDCT), which include, but are not limited to, the 13 forms of Ehlers–Danlos syndrome (EDS), generalised hypermobility spectrum disorder (G-HSD), Loeys–Dietz and Marfan syndromes and osteogenesis imperfecta. The most prevalent are the hypermobility form of EDS (hEDS) and G-HSD.3,4 Prior to 2017, hEDS and G-HSD were diagnosed as EDS–Hypermobility Type (EDS-HT) and joint hypermobility syndrome (JHS) respectively and, prior to this, as EDS-III and benign JHS.5 Of all patients with HDCTs, those with undiagnosed hEDS/G-HSD are the most likely to present to a general practitioner (GP).
Phenotypical presentation of hypermobility syndromes
The diagnosis of hypermobility syndromes is often delayed,6 as the phenotypical presentation of children/adolescents is widely variable.7,8 The child (and parent) may have sought GP consultations for various seemingly unrelated health issues. These include gastric upset fluctuating between nausea, diarrhoea and constipation; debilitating fatigue that is atypical for the child; orthostatic intolerance; allergies; and food and chemical sensitivities.7,8 The most common childhood presentations are joint sprains/subluxations/dislocations, persistent musculoskeletal pain, functional gastrointestinal disorders, urinary incontinence and anxiety. Additionally, parents may report behavioural issues and delayed motor milestones affecting activities of daily life and school functioning.3,9 The thread that connects these seemingly unrelated issues may be the most abundant tissue in the body: the connective tissue.4
Currently there is no confirmatory genetic testing for hEDS/G-HSD, likely as a result of multigene heterogeneity,3 so the diagnosis is clinical.4 It is essential for the primary practitioner to exclude other conditions with greater risk of severe acute health events. Twelve of the 13 forms of EDS are considered rare;5 some forms are associated with early mortality due to cardiovascular involvement and severe vascular fragility (ie vascular EDS, cardiac–valvular EDS and Marfan and Loeys–Dietz syndromes). Suspicion of cardiovascular involvement constitutes a trigger for urgent non-GP specialist referral, likely for echocardiography and molecular genetic testing.10
Presentations of each of the associated diagnoses require active investigation for other causes as per the appropriate clinical pathways. For example, debilitating fatigue would require exclusion of iron deficiency anaemia, while postural intolerance and/or syncope requires appropriate investigation to rule out cardiac causes. The ‘red flag’ triggers for further investigation or non-GP specialist referral for assessment of the underlying cause of the hypermobility are listed in Table 1. These include the patient’s signs and relevant information from their medical and family history that warrants further investigation for serious HDCT-related pathology including, but not limited to, severe forms of EDS, Marfan and Loeys–Dietz syndromes and osteogenesis imperfecta.
| Table 1. Indications for further investigation or non–general practitioner specialist referral in the presence of joint hypermobility |
| Patient |
Patient’s history |
Family history |
- Blue sclera
- Tall or short stature
- Cardiac murmurs
- Skin fragility
- Lens dislocation
- High myopia
- Ectopic lentis
- Developmental disability
- Intellectual disability
- Dysmorphic features
- Cleft palate/bifid uvula
- Multiple congenital anomalies
- Marfanoid body habitus
|
- Cardiac valve disease
- Congenital heart disease
- Vascular rupture or dissection
- Low-force fractures (two in children)
- Talipes equinovarus
- Severe scoliosis
- Wide atrophic scars
- Poor wound healing
- Aortic dilatation on echocardiography
- Recurrent hernia
- Organ rupture
- Recurrent pneumothoracies
|
- Cardiac valve disease
- Congenital heart disease
- Vascular rupture or dissection
- Mitral valve prolapse
- Sudden unexplained death
- Low-force fractures (three in adults)
- Formally diagnosed early onset osteoporosis
- Aortic dilatation on echocardiography
- Organ rupture
- Recurrent pneumothoracies
|
The 2017 EDS International Classification sets diagnostic criteria for hEDS (www.ehlers-danlos.com/heds-diagnostic-checklist). Table 2 provides a summary of the criteria for clinical diagnosis of hypermobile, classical and vascular forms of EDS. These three types of EDS are inherited in the autosomal dominant pattern. Designed for adults, the hEDS criteria can be challenging to use in a paediatric population with an evolving phenotype in whom typical recurrent hernias, stretch marks and atrophic scarring have not yet manifested. The recommended approach is to identify joint hypermobility and other signs and symptoms and to initiate targeted management while continuing to monitor and re-evaluate the children as they grow. Some children diagnosed with G-HSD will lose their hypermobility over childhood, whereas others will meet the hEDS criteria later in life. Further hindering hEDS diagnosis, the current criterion of a Beighton score ≥6/9 underestimates the presence of GJH in all males and in females aged >8 years.1
| Table 2. Summary of 2017 International Classification clinical criteria for diagnosis of hypermobile, classical and vascular Ehlers–Danlos syndrome (EDS)4 |
| |
Hypermobility EDS (hEDS) |
Classical EDS (cEDS) |
Vascular EDS (vEDS) |
| Major criteria |
Criterion 1
- Beighton score for GJH ≥6/9 if prepubertal; ≥5 if aged <50 years; ≥4 if aged ≥50 years.
If 1 point below Beighton cut-off, also need ≥2/5 historical GJH (‘Could you now or ever …’)
- Place hands on floor not bending knees?
- Bend your thumb to touch your forearm?
- Contort your body or do the splits?
- Dislocate your shoulder or kneecap on ≥1 occasion?
- Consider yourself as ‘double jointed’?
Criterion 2 (≥2 of A, B and C)
A. ≥5/12 of the following
- Soft/velvety skin
- Skin hyperextensibility
- Unexplained stretchmarks
- Atrophic scarring
- Piezogenic heel papules
- >1 abdominal hernia
- Unexplained urogenital prolapse
- Dental crowding or high/narrow palate
- Arachnodactyly (bilateral Walker or Steinberg signs)
- Armspan:height ≥1.05
- Mitral valve prolapse
- Aortic root dilatation >+2
B. Positive family history for hEDS (1° relative)
C. ≥1 of the following
- MSK pain in ≥2 limbs daily for ≥3 months
- Chronic widespread pain ≥3 months
- Recurrent dislocations (non-traumatic)
Criterion 3 (all three must be met)
- Absence of skin fragility
- Exclude other HDCT/automimmune and rheumatological conditions
- Exclude other hypermobility syndromes
|
- Skin hyperextensibility and atrophic scarring
- Meets Beighton cut-off scores for GJH as per hEDS
|
- Family history of vEDS (documented causal variant in COL31A)
- Arterial rupture at a young age
- Spontaneous sigmoid colon perforation in the absence of diverticular/bowel pathology
- Uterine rupture during pregnancy
- Carotid-cavernous sinus fistula formation in the absence of trauma
|
| Minor criteria |
- Easy bruising
- Soft, doughy skin
- Skin fragility (or traumatic splitting)
- Molluscoid pseudotumors
- Subcutaneous spheroids
- Hernia (or history of)
- Epicanthal folds
- Complications of GJH (sprains, joint pain, subluxation, dislocation, flexible flatfoot)
- Family history of 1° relative meeting clinical criteria
|
- Atraumatic (very easy) atypical bruising (eg cheeks/back)
- Thin, translucent skin with visible veins
- Characteristic facial appearance (large eyes, small chin, sunken cheeks, thin nose and lips, lobeless ears)
- Spontaneous pneumothorax
- Acrogeria
- Talipes equinovarus
- Congenital hip dislocation
- Hypermobility of small joints
- Tendon and muscle tears
- Keratoconus
- Gingival recession and fragility
- Early onset varicose veins (<30 years of age and nulliparous)
|
| Minimal criteria to meet diagnosis |
See above for 3 sets of criteria. The clinical diagnosis of hEDS needs the simultaneous presence of all criteria (1, 2 and 3).
|
Skin hyperextensibility and atrophic scarring, plus GJH and/or ≥3 minor criteria
|
A family history of vEDS, arterial rupture or dissection in people aged ≤40 years, unexplained sigmoid colon rupture, or spontaneous pneumothorax in the presence of other features of vEDS. Consider testing in the presence of a combination of the minor features. |
| Genetic testing |
No (research underway to identify likely multigene causation) |
Yes (most commonly COL5A1, COL5A2) |
Yes (commonly COL31A) |
| GJH, generalised joint hypermobility; HDCT, hereditary disorders of connective tissue; MSK, musculoskeletal |
The benefits of timely diagnosis
Diagnostic delay has serious consequences for quality of life, financial loss, unnecessary investigations, suboptimal therapies and worsening symptoms and disability.6,11 Patients report poor control over pain; disruption of activities of daily living and schooling; and suboptimal levels of physical activity.9 Persistent pain, unstable joints and dysautonomia may lead to kinesiophobia and consequently reduction of physical activity, which in turn exacerbates fatigue. Allocation of a provisional diagnosis and validation of their symptoms allows the child and their parents to focus on management, circumventing this vicious cycle.12
Management
The GP is key to comprehensively managing a patient with symptomatic hypermobile/hEDS/HSD as their central health coordinator. Management is multidisciplinary, shared with allied health practitioners and relevant specialists.13 The child’s treatment will vary depending on their presenting signs and symptoms, which will change over the course of their life.3,12 Asking the child and family to prioritise concerns as well as focusing on immediate functional deficits is a useful guide to making joint decisions about management priorities. Attention to education and empowerment of the patient and family are essential as part of the development of effective self-management strategies (Box 1).14
| Box 1. Strategies for managing hypermobility syndromes: Recommendations for patients/parents |
- Consult an experienced physiotherapist for targeted exercises for symptomatic joints and advice on appropriate sports to participate in and graded physical activity. Hypermobile joints are less likely to be unstable if the muscles that span them are strong and well controlled.
- Consult physiotherapists and occupational therapists to assist with taping, bracing, judicious stretching of tight muscles, pain education, pacing strategies, home modifications and ergonomics, and activities of daily living (eg handwriting, dressing, using cutlery, schoolwork).
- Develop coping strategies when symptoms are not overwhelming (eg have a medication plan for controlling pain before central sensitivity increases).
- Recognise and take advantage of the child’s most physically or cognitively alert times to engage in physically or mentally challenging activities.
- Seek the expertise of a dietitian if nutrition is not optimal (eg food intolerances,
under/overweight, excessive fatigue, gastrointestinal upset).
- Gaining knowledge and useful tips may assist when managing pain is a priority. The Agency for Clinical Innovation has a multi-session program that is targeted for the child and their family (www.aci.health.nsw.gov.au/chronic-pain/painbytes).
- Visit the Ehlers–Danlos Society website, which contains resources and a worldwide Healthcare Professionals Directory if the child is diagnosed with a form of Ehlers–Danlos syndrome (www.ehlers-danlos.com).
|
Musculoskeletal and other pain
Younger children with hypermobility syndromes present with musculoskeletal pain most frequently, but abdominal pain and headaches also occur.14 The GP can provide critical advice, such as using simple analgesics and modalities such as heat, ice and compression; using nonsteroidal anti-inflammatory drugs with caution (not in patients with upper gastrointestinal symptoms); and avoiding opiates. When pain is persistent and causes distress or limits function, psychological intervention to explore stressors and develop coping strategies for chronic pain, anxiety and related fatigue is helpful. Significant pain that interferes with daily activities and is refractory to regular therapies warrants referral to a paediatric pain specialist or paediatric pain services.14–16
Joint instability
Recurrent joint sprains, subluxations and low-force dislocations can be addressed by a physiotherapist with judicious use of supervised, progressive, motor control, strengthening and endurance exercise commencing in the normal physiological joint range and gradually moving into the child’s hypermobile range.17,18 Paced and graded activity programs establish a pattern of essential activities, including school and household tasks, before sports and recreation are added in a stepwise fashion.14 Bracing, splinting and taping are sometimes used to assist with the child’s exercise program and functional performance, which also focuses on training balance and proprioception. Consultation with a podiatrist may assist with footwear advice and orthoses to address overpronation and improve lower limb alignment.19 Children and their parents commonly report finger, hand and wrist pain and instability affecting fine-motor and handwriting ability with consequent impact on academic performance and activities of daily living.13 These issues should be assessed and treated by a paediatric occupational therapist.
Gastroenterological and urological involvement
Most paediatricians are familiar with functional gastrointestinal disorders and can advise on symptom management as needed. There are emerging therapies and strategies for symptom management, particularly dietary strategies and a multidisciplinary approach to chronic symptoms.20–22 Urinary incontinence, present in 26% of a sample of 89 Australian children with JHS/EDS-HT, significantly affects quality of life and warrants management.23
Fatigue
Debilitating fatigue is a common complaint that affects a child’s social, educational and recreational life. Poor sleep, muscle weakness/debilitation and dysautonomia have all been shown to be associated with worse fatigue in JHS.14,24 Graded reconditioning, hydrotherapy, cognitive behavioural therapy, pacing and sleep hygiene may assist with fatigue and chronic pain.
Dysautonomia
Some children present with dizziness, weakness and/or palpitations on standing or after exercise or bathing; suspicion of the presence of postural orthostatic tachycardia syndrome should facilitate non-invasive investigation (usually with a tilt test and beat-to-beat haemodynamic monitoring).25 There is some evidence of the benefit of ingested isotonic drinks, intravenously administered saline in a hospital setting,26 medications,27 dietary modifications, aerobic exercise,28 use of pressure garments,29 breathing techniques and active tensing of lower limb muscles prior to standing to alleviate symptoms.30 Debilitating postural intolerance and/or
faints may require evaluation by a paediatric cardiologist.
Psychological disorders
The prevalence of anxiety disorders in those with hEDS/G-HSD is high, and identifying and addressing early signs may prevent worsening with age.31 Where the possibility of self-harm exists, differential diagnosis is imperative.32 Paediatric psychological and psychiatric referral may complement other allied health management.
When consulting with children with syndromic hypermobility, the first challenge for the primary practitioner is connecting the common thread that links the signs and symptoms. Recognising the triggers for further investigation or non-GP specialist referral is essential to exclude serious disease. Using a personalised medicine approach, recognising and addressing patient and parent priorities, which change as the child matures, will assist in targeted management.
Key points
- The presentation of hypermobility syndromes is highly heterogeneous, the only consistent feature being the presence of general or peripheral joint hypermobility.
- The role of the GP is vital in assisting patients and their parents to identify short- and longer-term assessment/diagnostic and management priorities
- Optimal care requires navigation of the complex array and interplay of associated medical conditions, necessitating multidisciplinary coordinated management.
- Early recognition and proactive appropriate management in childhood may reduce the impact of this debilitating condition in later life.