Case
A male engineer aged 47 years attended his general practice for blood test results. At the consultation, multiple pinhead-sized flat red spots were noticed on his fingers and face (Figure 1).
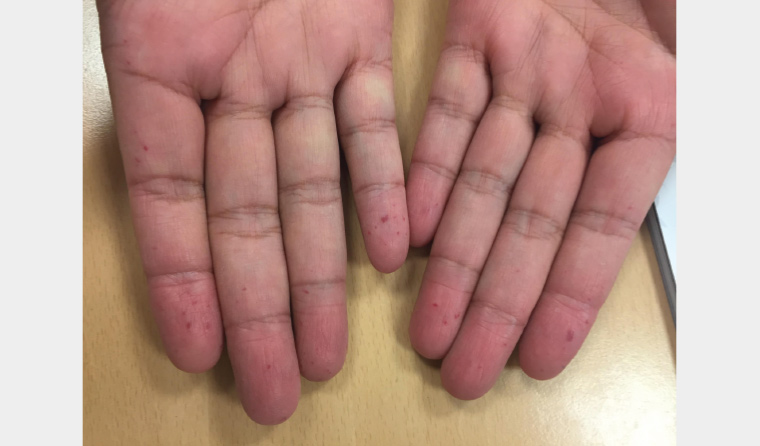
Figure 1. Multiple pinhead-sized flat red spots on the patient’s hands
The patient also reported near-daily nosebleeds from three years of age, which subsided with local pressure. The skin lesions had been present for nine years.
His medical history included essential hypertension (nine years), a rash to sulfonamide medications and a family history of diabetes. He did not smoke or drink alcohol. He was married and had a son and daughter.
He had no known relatives with similar skin lesions but a strong family history of cerebral haemorrhage (father at 62 years of age, brother at 44 years of age, paternal grandfather at 65 years of age and son at 16 years of age). He had no melena or bleeding symptoms from other sites.
Examination of the skin revealed blanching lesions on the fingertips, face, ears and upper neck. Oral and ear, nose and throat (ENT) examinations were normal, with no obvious bleeding points.
His blood pressure was 111/66 mmHg, and his body mass index was 27.8 kg/m2.
His results included haemoglobin 78 g/L (reference range 130–170 g/L), microcytic anaemia, glycated haemoglobin 41 mmol/mol (5.9%), and normal lipids and liver function tests. Iron studies showed iron 3.61 µmol/L (reference range 5.83–34.50 µmol/L), total iron binding capacity 68 µmol/L (reference range 45–80 µmol/L) and iron saturation 5% (reference range 15–45%). International normalised ratio, activated partial thromboplastin time and platelet count were normal.
Question 1
What are these lesions? What is the difference between telangiectases, arteriovenous malformations (AVMs) and purpura?
Question 2
What is this disease, and what conditions are associated with this disease?
Question 3
What are the genetics of this disease?
Answer 1
These lesions are telangiectases, which are visible, dilated small blood vessels. They can be arterioles, capillaries or venules. They blanch with light pressure. In contrast, purpura, or mucocutaneous bleeding, are classified as petechiae (<2 mm) or ecchymoses (≥2 mm) and do not blanch to pressure.1 AVMs are communications between the arterial and venous circulation without normal intervening capillaries.2
Answer 2
The combination of recurrent epistaxis, iron deficiency anaemia and mucocutaneous telangiectases suggest hereditary haemorrhagic telangiectasia (HHT; also called Osler–Weber–Rendu disease), an autosomal dominant condition affecting approximately one in 5000 people.3 Mucocutaneous telangiectases and solid organ AVMs occur, with age-related penetrance.
The Curaçao criteria4 aid clinical diagnosis and allow identification of this condition (with genetic testing performed in select circumstances).3 Four criteria are identified: spontaneous and recurrent epistaxis, multiple mucocutaneous telangiectases in characteristic areas (lips, oral cavity, nose, fingers), visceral involvement (gastrointestinal, liver, lung, central nervous system) and a first-degree family member with HHT. Three or more features are diagnostic of HHT; two features make HHT possible/suspected, and no features or one feature make HHT unlikely but still possible in a child.4 A negative gene test does not exclude the diagnosis.
Prevalence of lesions increases with age and is (approximately): cutaneous telangiectasia (75–90%), epistaxis (78–96%), gastrointestinal (75–80%), hepatic (32–78%), pulmonary (15–60%), cerebral (10–23%) and spinal (1%) AVMs.2,4
Answer 3
This disorder involves mutations of ENG or ACVRL1 genes in 85–96% of cases and several other genes infrequently. SMAD4 is associated with both HHT and juvenile polyposis syndrome.4,5 Relatives should be counselled regarding genetic testing if the index case has a recognised gene mutation.3
Case continued
The patient was referred for ENT, genetics and internal medicine consultations for suspected HHT.
His epistaxis was treated with moisturising topical therapies to keep the nasal mucosa moist.
After counselling, he underwent genetic testing, and a mutation of ACVRL1 was identified, leading to a formal diagnosis of HHT.
Question 4
What management interventions would be helpful?
Answer 4
Pulmonary AVMs should be sought on diagnosis, and antibiotic prophylaxis is needed for surgical/dental procedures for patients with pulmonary AVMs because of the risk of septic emboli. Pulmonary AVMs result in a direct right-to-left shunt with impairment of the filtering capabilities of the lung, permitting the passage of emboli and bacteria through the capillary bed, potentially causing transient ischaemic attacks, strokes or brain abscesses.
The patient should receive oral iron replacement for anaemia. Gastroscopy should be performed if the anaemia does not respond to iron supplementation. Patients with the SMAD4 variant should undergo colonoscopy.
Liver AVMs may be unrelated to HHT but should be investigated further if complicated by other features (eg heart failure, pulmonary hypertension, abnormal liver function tests, abdominal pain).3
Internal AVMs can lead to increased morbidity/mortality during pregnancy, so pregnant patients with HHT should generally be managed in tertiary care, especially if pulmonary or cerebral AVMs are present. Vaginal delivery and/or epidural analgesia is still possible in select cases.3
A website for patients and physicians is available (https://curehht.org).6
Conclusion
Following treatment with an iron supplement, the patient’s anaemia resolved. Imaging showed no evidence of cerebral, pulmonary or hepatic AVMs.
His son was also found to have the ACVRL1 mutation, while his daughter was negative.
During this time, the patient’s body weight continued to rise, and he developed diabetes.
Key points
- At a prevalence of one in 5000 people, HHT is not a rare disease.
- Clinical diagnosis is based on the Curaçao criteria.
- A formal diagnosis allows screening for internal complications and early intervention.