CASE
A man, aged 74 years, presented with a four-month history of fatigue, breathlessness and malaise. Aside from left auricular pain, the patient denied any focal symptomatology, including headaches, visual changes, jaw claudication and abdominal pain. There was no fever or weight loss. His past medical history included type 2 diabetes and an unprovoked left lower limb deep venous thrombosis three months prior. The patient was previously well.
On examination, a faint macular skin rash affecting the torso and limbs was noted. Rheumatological assessment showed no synovitis, joint deformities or nailfold vasculitis. There was no bony tenderness, lymphadenopathy or hepatosplenomegaly.
Routine bloods revealed a macrocytic anaemia (haemoglobin 78 g/L [normal range: 125–175g/L], mean corpuscular volume 108 fL [80–99 fL]. Previous haemoglobin had been 128 g/L 11 months prior to presentation and 99 g/L seven months prior to presentation). Inflammatory markers were elevated (C-reactive protein [CRP] 97 mg/L [<5 mg/L], erythrocyte sedimentation rate 135 mm/h [<20 mm/h]). Active B12 was 75 pmol/L (>35 pmol/L) and serum folate was 13.2 nmol/L (7–40 nmol/L). The platelet count was 344 × 109/L (150–450 × 109/L) and the white cell count was 9.7 × 109/L (4.0–11.0 × 109/L).
Systemic lupus erythematosus as a differential diagnosis was considered, but autoimmune serology, including rheumatoid factor, anti-cyclic citrullinated peptide, antinuclear antibody, extractable nuclear antigen and antineutrophil cytoplasmic antibodies were negative, as was a haemolytic screen.
A whole-body computed tomography (CT) scan excluded solid malignancy. Positron emission tomography showed no evidence of large vessel vasculitis or fluorodeoxyglucose-avid malignancy.
Given the patient’s chronic inflammatory symptoms, auricular pain, previous thromboembolism and haematological abnormalities, the possibility of VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome was ultimately considered. The patient was commenced on oral prednisolone (50 mg/day).
Bone marrow biopsy showed hypercellular marrow with increased granulopoiesis and bilineage dysplasia involving erythropoiesis and megakaryopoiesis (in <10% of cells) consistent with an evolving myelodysplastic disorder. Very occasional myeloid precursors with vacuolated cytoplasm were identified (Figure 1).
Lower limb Doppler ultrasound revealed bilateral non-occlusive thrombi in the superficial femoral veins. Progress CT of the chest showed extensive pulmonary infiltrates (Figure 2). Molecular genetic testing was positive for a ubiquitin-like modifier activating enzyme 1 (UBA1) variant. The patient was subsequently diagnosed with VEXAS syndrome.
The patient commenced treatment for VEXAS syndrome, comprising a weaning dose of prednisolone and subcutaneous tocilizumab (162 mg weekly). After six months of tocilizumab, the patient’s rash and auricular pain had resolved. Haemoglobin was 119 g/L, CRP was 0.4 mg/L and the erythrocyte sedimentation rate 9 mm/h.
At the time of writing, the patient remains well systemically and continues to undertake weekly tocilizumab injections. Apart from postinflammatory scarring, a repeat CT scan of the chest showed resolution of pulmonary infiltrates. There has been no flare of disease.
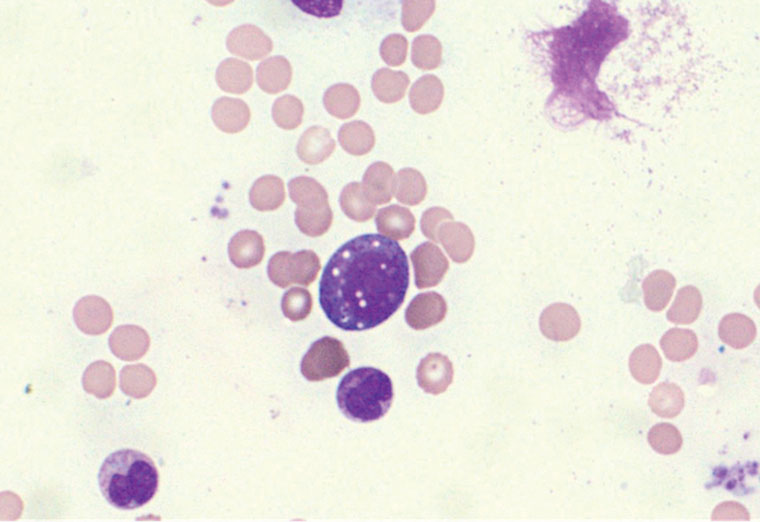 Figure 1. Bone marrow aspirate demonstrating cytoplasmic vacuolation of myeloid precursor cells.
Figure 1. Bone marrow aspirate demonstrating cytoplasmic vacuolation of myeloid precursor cells.
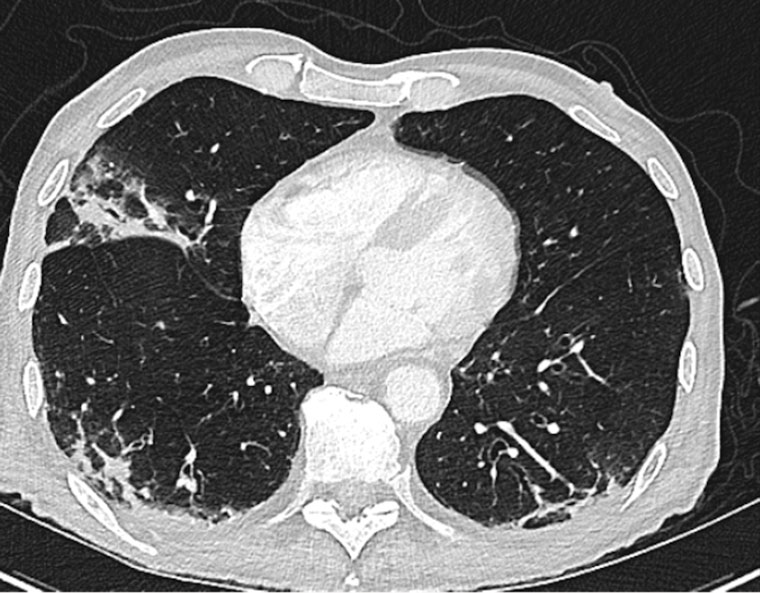 Figure 2. Computed tomography scan of the chest showing extensive lung parenchymal abnormality with subpleural opacification and associated traction dilatation.
Figure 2. Computed tomography scan of the chest showing extensive lung parenchymal abnormality with subpleural opacification and associated traction dilatation.
Question 1
What is VEXAS syndrome?
Question 2
What are the clinical features of VEXAS syndrome?
Question 3
How is VEXAS syndrome diagnosed?
Question 4
What treatment is available for VEXAS syndrome?
Answer 1
First described by Beck et al1 in 2020, VEXAS syndrome is a novel multisystem haematoinflammatory disease associated with disorders of the bone marrow. It is caused by an acquired mutation in UBA1, an X-linked gene encoding ubiquitin-activating enzyme 1 (E1), which is integral to intracellular signalling and protein degradation.
Although VEXAS syndrome is a rare disease with an estimated prevalence of 1 in 14,000,2 there have been increasing diagnoses in retrospective genetic testing, suggesting the syndrome might be more common than initially thought.3
Answer 2
VEXAS syndrome most commonly affects males. Symptoms typically occur in the sixth decade.1,3,4 The severity of VEXAS syndrome ranges from mild to life-threatening.
Constitutional symptoms, such as fatigue, fever, night sweats and weight loss, affect almost all patients.1,2 Dermatological manifestations are frequently reported, including nodules, tender plaques and neutrophilic dermatosis, although a skin biopsy was not performed in this case. Lung involvement presents as pulmonary infiltrates and pleural effusions. Other clinical features include nose and ear chondritis, arthralgias and thromboembolic disease.1,4,5 Venous thromboembolism, demonstrated in up to 40% of patients, is typically unprovoked and occurs within two years of inflammatory symptom onset,2 consistent with the case presented.
Clinical presentation between patients is diverse due to the ubiquitous nature of E1, making VEXAS syndrome difficult to recognise, accurately diagnose and treat. Patients often meet classification criteria for other autoimmune and inflammatory conditions, such as Sweet’s syndrome, relapsing polychondritis and polyarteritis nodosa.1,2 Consequently, symptoms might be present for several years before a diagnosis of VEXAS syndrome is made.
The haematological abnormalities most commonly observed in VEXAS syndrome are macrocytic anaemia, lymphopenia, thrombocytopenia and monocytopenia. Patients might concurrently have a myelodysplastic syndrome or plasma cell dyscrasia, such as a monoclonal gammopathy of unknown significance.1 Because VEXAS syndrome is a progressive disease, major complications contributing to disease morbidity and mortality relate to myelodysplasia and the increased risk of transformation into haematological malignancies, such as acute myeloid leukaemia.
Answer 3
Diagnosis of VEXAS syndrome requires bone marrow biopsy, demonstrating characteristic vacuolisation of myeloid precursor cells, and genetic testing confirming loss of UBA1 function.1,5
Answer 4
Because VEXAS syndrome is a novel entity, there is no established management guideline. Current treatment recommendations have been based on observational studies and case series.2 A recent systematic review, involving 36 publications and 116 patients, concluded that there is insufficient available evidence to support one standardised treatment paradigm.4
In this case, tocilizumab, an interleukin (IL)-6 inhibitor, was the preferred treatment based on the significantly elevated IL-6 and CRP levels observed in VEXAS patients.2,5 Tocilizumab was well tolerated without any adverse effects.
Other described treatment options for VEXAS syndrome include Janus kinase inhibitors (ruxolitinib), hypomethylating agents (azacitidine), anti-IL-1 therapy (anakinra), glucocorticoid monotherapy and allogenic stem cell transplantation.3,4 Treatment for VEXAS syndrome should be individualised such that patient factors and disease phenotype are considered. For example, hypomethylating agents are suggested for patients with concomitant myelodysplastic syndrome. To date, overall literature on the optimal treatment of VEXAS syndrome remains inconclusive.1,4,5
Key points
- VEXAS syndrome is a newly described multisystem autoinflammatory disease caused by a somatic mutation in the UBA1 gene.
- VEXAS syndrome is a differential to consider in a male patient with persistent, unexplained, treatment-refractory inflammatory manifestations with cytopenias.
- Although the optimal treatment for VEXAS syndrome remains unclear, good clinical and serological outcomes can be achieved with combination steroid and disease-modifying anti-rheumatic drug therapy.