Illustrative cases
Case 1. A young sudden cardiac arrest survivor
A previously well girl, aged 11 years, finished her day at school and was running to the train station. She had a cardiac arrest within the station near a pharmacy. Cardiopulmonary resuscitation (CPR) was commenced. An ambulance was called and arrived to find her in ventricular fibrillation, which reverted to sinus rhythm with one shock. She survived the event with no neurological effects, and subsequent echocardiography identified hypertrophic cardiomyopathy (HCM); an implantable cardioverter defibrillator (ICD) was implanted. DNA testing revealed a pathogenic mutation in the most common HCM gene, MYBPC3. Subsequent family screening of first-degree relatives identified both her father and one of the brothers to have HCM and carry the gene mutation, and appropriate prevention strategies including insertion of an ICD were initiated. Her two other brothers were shown to not carry the gene mutation and so were released from lifelong clinical surveillance.
Case 2. A young sudden cardiac death and subsequent investigation
A woman aged 22 years with a history of unexplained syncope 12 months prior had a sudden cardiac arrest while having an argument with her father. Despite all resuscitative efforts, the woman did not survive. Subsequent comprehensive post-mortem review failed to find a cause of death, and the case was considered as a sudden unexplained death (SUD). Clinical review of the family revealed her mother to have a prolonged QT interval at 500 msec and one of her sisters to have a borderline QT interval of 460 msec. A ‘molecular autopsy’ (genetic analysis) was performed on DNA extracted from a blood sample of the decedent collected at post-mortem and identified a pathogenic mutation in the KCNH2 gene known to cause familial long QT syndrome (LQTS). This gene result provided a cause of death for the family, and cascade genetic testing of other family members could be initiated to identify at-risk relatives. The mother carried the gene mutation and was commenced on beta-blockers for arrhythmia prevention, while the sister with the borderline QT interval did not carry the gene mutation and so was released from lifelong clinical surveillance.
Sudden cardiac death in the young
The sudden death of a young person is a rare but tragic event that can have a devastating impact on both the individual’s family and the wider community. Sudden non-traumatic deaths result from a variety of disorders (including asthma, pulmonary embolism, epilepsy and intracranial haemorrhage), but in people aged ≤35 years, up to 56% are attributed to cardiac disorders.1,2 Sudden cardiac death (SCD) is an unexpected death from a cardiac cause in a previously healthy individual. By definition, SCD occurs within one hour of symptom onset (or within 24 hours of the individual last being seen in good health if the death is unwitnessed), and it is a complication of a number of genetic and acquired cardiovascular conditions.3 Although 75% of individuals who experience SCD are asymptomatic, one study showed that up to 8% had seen a physician for syncope or seizure prior to death.4
The annual incidence of SCD in the young in Australia and New Zealand is reported to be 1.3 cases per 100,000,5 but this figure is likely an underestimate given that a proportion of traumatic deaths (eg drownings or motor vehicle accidents) are likely to be the consequence of cardiovascular collapse due to a primary cardiac disorder. SCD is more common in young men than in young woman, with a gender ratio of nearly 3:1.5 Although sudden deaths of young sportsmen and sportswomen during competition are more commonly seen in the media, the majority of SCD events in young people occur during sleep or in periods of rest (Figure 1).5,6
Establishing a cause of death in young people who die suddenly is critical for providing optimal care to the bereaved family, and therefore a comprehensive post-mortem examination is of utmost importance. A premorbid history and a detailed autopsy including macroscopic and histologic assessment, as well as toxicology screening, can identify non-cardiac causes of death and structural cardiac abnormalities (including anomalous coronary artery anatomy, cardiomyopathy, premature coronary artery disease and valvular pathology with or without great vessel involvement) that could explain the sudden death. SCDs where no cause is identified after complete post-mortem examination are deemed sudden unexplained deaths (SUDs) and are presumed to be the result of primary arrhythmogenic disorders.6 Young women are more likely to experience SUD than explained SCD, and SUD is more likely to occur during sleep.5 SUD accounts for 40% of all SCDs in individuals aged 1–35 years, while the proportion of SCD with a cause identified on autopsy increases with age; only 24% of deaths in the 31–35 years of age cohort are considered unexplained.5
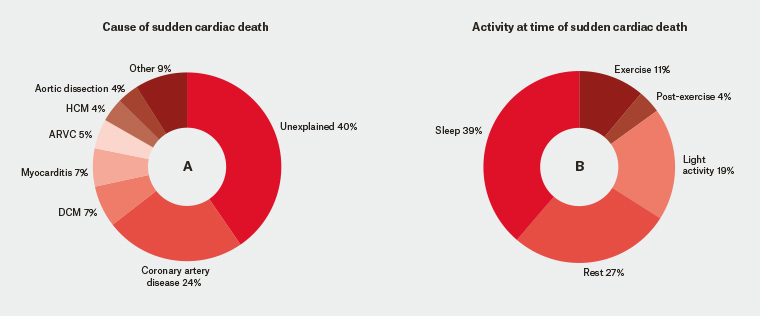
Figure 1. Sudden cardiac death in young Australians
Cause of sudden cardiac death (Chart A) and activity at time of sudden cardiac death (Chart B) in young Australians.5
ARVC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy
Genetic causes of SCD in the young
In those aged over 35 years, coronary artery disease is the most common cause of SCD and is present in >80% of cases, but in young people, genetic cardiac conditions including inherited cardiomyopathies and primary arrhythmia syndromes are more common (Figure 1).7 The inherited cardiomyopathies typically display structural abnormalities that are identifiable on autopsy either macroscopically or histologically. The inherited arrhythmia syndromes result from mutations in genes that encode cardiac ion channel function and predispose to malignant tachyarrhythmias at the molecular level with no discernible abnormalities on routine post-mortem examination. Although these conditions can all have the devastating consequence of SCD, they have distinct clinical features, genetic aetiologies and management strategies. These are summarised in Table 1.
| Table 1. Genetic causes of sudden cardiac death (SCD) |
| Condition |
Clinical features on screening |
Autopsy findings |
Key gene associations |
Management options for prevention of SCD |
| HCM |
Left ventricular wall thickness ≥15 mm measured by any imaging modality |
Increased left ventricular thickness
Myofibrillar disarray and interstitial fibrosis16 |
MYBPC3 (30–40%), MYH7 (20–30%), TNNT2 (10%), TNNI3 (7%)17 |
ICD in selected patients
Exercise restriction in some cases |
| ARVC |
Epsilon wave or T-wave inversion in right praecordial leads (V1, V2 or V3)18
Impaired right ventricular function on cardiac imaging |
Fibrofatty replacement of the ventricular myocardium |
PKP2 (46%), PLN (5%)17 |
Exercise restriction
ICD |
| DCM |
Increased left ventricle cavity size with impairment of systolic function on cardiac imaging (not attributable to other causes such as coronary artery disease, alcohol, cardiotoxic chemotherapy) |
Increased cardiac mass with dilated ventricles and no inflammatory myocardial or coronary artery disease16 |
TTN (25%)17
LMNA17
DES |
ICD in selected patients |
| LQTS |
ECG: QT interval duration exceeding normal value (>440 ms in men and >460 ms women)
LQTS1: events during exertion (eg swimming)
LQTS2: events during sleep/rest
LQTS3: events provoked by loud noise |
Nil |
KCNQ1 (40–45%), KCNH2 (30–35%), SCN5A gain of function mutation (10%)19 |
Drug avoidance –
www.crediblemeds.org
Beta blockers (eg propranolol or nadolol)
Sodium channel blockers – mutation dependant use in LQTS319
Left cardiac sympathetic denervation20 |
| CPVT |
Stress ECG: bidirectional or polymorphic ventricular tachycardia |
Nil |
RyR2 (50–65%)17,19
CASQ2 (<5%)17 |
Beta blockers
Exercise restriction
Left cardiac sympathetic denervation
ICD is therapy of last resort |
| Brugada syndrome |
≥2 mm ST-segment elevation with coved-type morphology in ≥1 lead of the right precordial leads (V1–V3 in 4th–2nd intercostal spaces)
Spontaneous
Drug challenge with sodium channel blocker
Event triggers include sleep/rest and fever |
Nil |
Genetically heterogeneous condition
SCN5A loss of function mutation (20–25%)
|
Fever management
Drug avoidance –
www.brugada-drugs.org
Quinidine21–23
ICD |
| SQTS |
ECG: QT interval duration <330 msec or <360 with high-risk features24 |
Nil |
KCNH2, KCNQ1, KCNJ2
Gene mutation identified in only 14%24 |
ICD therapy |
| ARVC, arrhythmogenic right ventricular cardiomyopathy; CPTV, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; ECG, echocardiography; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter-defibrillator; LQTS, long QT syndrome; SQTS, short QT syndrome |
Genetic testing in SCD in the young
Genetic disorders associated with SCD are most commonly inherited in an autosomal dominant pattern, resulting in 50% of first-degree relatives (parents, siblings and children) inheriting the mutation in the disease gene. Identifying a genetic cause of SCD has major implications for the clinical evaluation of the survivor of sudden cardiac arrest and their family (Case 1), or families where SCD has occurred (Case 2). When a cause of death that is known to have a genetic basis (eg HCM) has been identified on autopsy, targeted genetic testing is performed to identify mutations in genes associated with the condition. In cases of SUD (autopsy-negative SCD), DNA extracted from post-mortem blood samples or tissue can be tested to identify occult genetic causes of arrhythmogenic death. This is called the ‘molecular autopsy’.
The molecular autopsy seeks to identify an underlying aetiology in cases of previously unexplained SCD. The diagnostic yield of the molecular autopsy has increased with the rapid advances in genetic technologies. Since its inception in 2004,8 post-mortem genetic testing has moved from screening for a single gene mutation to gene panels with direct sequencing of the most common arrhythmia genes, including KCNQ1, KCNQ2 and SCN5A associated with LQTS, and RYR2 associated with catecholaminergic polymorphic ventricular tachycardia (CPVT), with detection rates thought to be 15–20%.3 With the development of next generation or ‘deep’ sequencing, more rapid and broader analysis of genomic information has led to an increased number of genes being included on SCD genetic panels. More recently, detection rates of >30% have been reported with whole exome sequencing,9 and the prospect of whole genome sequencing that provides coverage of introns and mitochondrial DNA is drawing closer.
Establishing the pathogenicity of a mutation is increasingly challenging with the vast amounts of genetic information now available. When a novel genetic variant is identified in an SCD proband, every effort is made to classify the genetic alteration as either a benign variant or a disease-causing mutation. Type of nucleotide variation (eg frame-shift, insertion, deletion, missense mutation, single nucleotide polymorphism), frequency of the mutation in the general population (established from genetic databases) and co-segregation of the variant within the family are among the features considered when assessing the likelihood that the variant could account for disease.10 This is particularly difficult in cases of SCD because the true phenotype of the proband is difficult to assess and genetic heart diseases typically display incomplete penetrance and variable expressivity.3,10 If a mutation fails to be classed as pathogenic or benign, it languishes as a ‘variant of unknown significance (VUS)’ and has no clinical use. Genetic tests are ‘non-binary’ and classifications are not finite; they exist on a spectrum and can be dynamic.11 Mutations are periodically reassessed and may be reclassified as new information becomes available as a result of research or clinical assessment.10 It is imperative that the probabilistic nature of genetic results is understood when interpreting genetic tests and communicating genetic information to families.11
Caring for families affected by SCD in the young
The sudden and unexpected death of a young person presents perhaps one of the most complex clinical situations faced by healthcare professionals, and the care of families affected by SCD and of survivors of sudden cardiac arrest is best delivered in a specialised, multidisciplinary genetic heart disease clinic.12 Management issues that need to be addressed include the diagnosis and genetic evaluation in the proband, clinical cardiovascular care of the survivor and family, and psychosocial support and counselling. The various professionals outlined in Figure 2 possess the diverse skills required to manage this myriad of clinical issues faced by survivors and families.
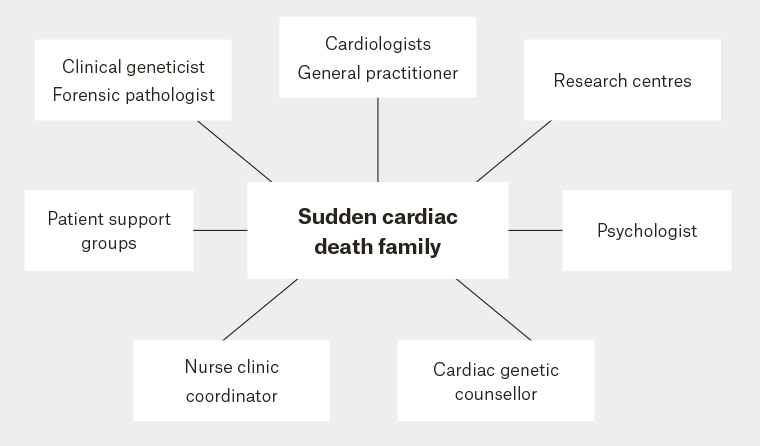
Figure 2. Family-centred model of multidisciplinary cardiac genetic care25
The key goal in the management of families affected by SCD is to prevent SCD in surviving relatives. Screening of first-degree relatives aims to identify those at risk and institute preventive therapies. A pre-screening discussion about the possible outcomes of clinical and genetic testing is required in all families and includes the implications of a positive diagnosis in terms of personal and life insurance. Clinical screening includes a detailed history and physical examination, electrocardiography, echocardiography and stress testing as well as adjunctive testing such as pharmacological provocation or cardiac magnetic resonance imaging where indicated.12 Identifying a pathogenic genetic mutation in a survivor of sudden cardiac arrest or an individual whose cause of death was SCD has major implications for family screening (Cases 1 and 2). When a genetic diagnosis has been made, evaluation of relatives for the known mutation (cascade testing) risk-stratifies family members into those with the mutation who will require intervention (eg implantable cardiac defibrillator, beta-blocker therapy or lifestyle modification) or ongoing follow-up, and those without the mutation who can confidently be released from follow-up. VUS must not be used for cascade testing, and families without a pathogenic mutation identified are to be followed up clinically.
A wide range of emotions may be experienced in response to the sudden and devastating loss of a loved one. In particular, surviving relatives have been shown to have symptoms of anxiety and depression. Mothers of individuals whose cause of death was SCD appear to be the most severely affected, with >50% reporting anxiety levels suggestive of an anxiety disorder.13 During the process of clinical and genetic evaluation, it is vital that the grief associated with SCD is acknowledged and the psychosocial wellbeing of families is prioritised. The genetic counsellor is pivotal in the provision of holistic family-centred care and develops a strong relationship with surviving relatives. In addition to ‘sensitively and effectively’ conveying complex genetic information to families, the genetic counsellor is often the key contact for families and can play an important part in normalising grief responses.14 This close bond can help identify those experiencing severe or prolonged grief who could benefit from specialised psychologist care as they try to come to terms with the sudden loss of a young relative.15
Implications for general practice
The general practitioner (GP) is at the coalface of SCD in the community and is an integral member of the multidisciplinary team caring for families affected by SCD and survivors of cardiac arrest. General practice occupies a unique position in the healthcare system where a single practitioner (or practice) frequently cares for multiple generations of a family and also has a close link to the community at large. These factors place the profession in the perfect position to respond to management of the family following SCD in the young. The key roles for the family doctor in the management of SCD include identifying at-risk patients and families who should be referred for cardiac assessment and providing long-term clinical and psychosocial care for families post-SCD in consultation with the genetic heart disease centre. The GP may also be the first to recognise the possibility of a genetic heart disease in an individual or in the family history, and should refer such patients for further cardiac assessment (Box 1). Basic information about genetic heart diseases and SCD in the young is available at various online sources (Box 2).
| Box 1. When to suspect genetic heart disease and consider cardiac assessment |
- Unexplained cardiac arrest or sudden death in a relative
- Diagnosis of a genetic heart disease in a relative
- Sudden infant death syndrome in a relative
- Unexplained syncope or any syncope on exertion or emotional excitement
- Seizures with normal neurological assessment
- Unexplained drownings or motor vehicle accidents
|
| Box 2. Resources for general practitioners and patients |
- Australian Genetic Heart Disease Registry, www.heartregistry.org.au
- Cardiac Society of Australia and New Zealand, www.csanz.edu.au/resources
- Position statements on genetic heart diseases including hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, dilated cardiomyopathy, long QT syndrome, catecholaminergic polymorphic ventricular tachycardia and Brugada syndrome
- Sudden Arrhythmic Death Syndrome Australia, www.sads.org.au
- Cardiomyopathy Association of Australia, www.cmaa.org.au
|