This article is the second in a series of articles on important topics in neurology.
The clinical course of spinal muscular atrophy (SMA) is changing in the era of disease-modifying therapies. What was once a fatal disease at the severe end of the spectrum is becoming a treatable condition with improved functional status and outcomes. Early recognition and prompt intervention are now known to improve clinical outcomes, including survival and acquisition of motor milestones. This places the primary care provider into a pivotal role for early suspicion, recognition and referral for SMA, considering that ‘time is motor neuron’. Ongoing care and follow-up will also be increasingly relevant as SMA becomes a chronic care condition requiring routine and disease-specific healthcare.
The aim of this article is to provide an overview of SMA for a primary healthcare setting in the context of a changing clinical course of disease in order to support early suspicion and referral for SMA, thus allowing affected children to access time-critical treatment. In addition, it summarises the novel therapies and important long-term management features pertinent to the general practitioner (GP) caring for these children and families.
What is spinal muscular atrophy?
SMA is an inherited autosomal recessive neuromuscular disorder characterised by progressive muscle weakness and atrophy in individuals who are cognitively normal. In the general population there is a carrier frequency of approximately one in 40 people, resulting in the condition affecting roughly one in 10,000 live births.1 Although a rare disease, untreated SMA is the leading genetic cause of death in children under two years of age.2
SMA is caused by a deficiency in the survival motor neuron (SMN) protein due to homozygous mutations in the survival motor neuron 1 (SMN1) gene. An almost identical nearby SMN2 gene can produce a small amount of functional SMN protein. SMN2 copy numbers vary between individuals, and the severity of SMA is largely, although not solely, inversely related to the number of copies of SMN2.3,4
How do children with spinal muscular atrophy present?
People with SMA are on a continuum of severity, from severe hypotonia and weakness to mild proximal muscle weakness and walking unassisted. Historically, SMA has been classified into four major phenotypes (three affecting children) that are separated by the age at symptom onset and maximal motor milestone achieved (Table 1).
| Table 1. A summary of spinal muscular atrophy (SMA) subtype classification and presentation2,4,7,23–25 |
| SMA type |
Incidence |
Age of onset |
Best motor milestone |
Untreated
life span |
Typical SMN2 copy number |
Presenting features |
1
|
~60% of cases |
0–6 months |
Never sits |
<2 years (without respiratory support) |
80% of patients have 1–2 copies |
Poor head control, hypotonia, absent tendon reflexes, ‘frog leg’ posture, tongue fasciculations, poor swallow, paradoxical breathing |
| 2 |
~27% of cases |
6–18 months |
Sits independently |
Most (~70%) alive at 25 years |
>80% of patients have three copies |
Absent tendon reflexes, scoliosis, low muscle tone, proximal weakness, motor delay with difficulty standing/walking, hand tremor |
| 3a |
~12% of cases |
>18 months |
Stands and walks |
Almost normal |
96% of patients have 3–4 copies |
Proximal weakness greater in legs than arms, waddling gait, trouble with stairs and rising from floor, hand tremor |
| 3b |
>3 years |
| 4 |
~1% of cases |
>21 years |
Normal |
Normal |
75% have four or more copies |
Adult-onset mild proximal weakness |
| SMN, survival motor neuron |
Symptoms and signs to look for and approach to examination
The classical clinical triad of SMA is muscle atrophy, fasciculations and areflexia in a cognitively normal child. However, diagnosis can be delayed because subtle early signs may be difficult to detect. For example, as the individual with SMA transitions from presymptomatic to symptomatic, preserved or brisk tendon reflexes may be evident on examination and fasciculations may not yet be present.5,6
Affected children experience a progressive, predominantly proximal muscle weakness, the presenting symptoms of which will be determined by severity of the disease and age of the child. In infants, severe weakness often results in an inability to obtain basic motor milestones, such as head control or rolling, and may result in swallowing difficulties causing growth failure.3,7 Feeding difficulties typically present after the first few weeks of life, and loss of rolling or head control milestones have been reported by parents of children in this age group.5 Muscles of the pelvic girdle are commonly affected early, and as a result of the combination of muscle weakness and hypotonia, hips will often be externally rotated with flexed knees, giving a ‘frog leg’ position. Minimal antigravity movement at the shoulder and significant head lag can be observed.3 There is usually relative sparing of the diaphragmatic muscles and, as a result, breathing can appear paradoxical, with intercostal recession and abdominal distension with inspiration.3
In older infants aged >6 months and children, the signs of weakness can be more subtle. Affected children will typically have an initially normal motor trajectory, including sitting independently. However, they then show difficulties relative to their peers, such as inability or delay in walking; trouble with activities that require proximal muscle groups such as rising from the floor, climbing and descending stairs; and frequent falling.5,8 Examination may show objective weakness in these muscle groups and possibly the presence of wasting, fasciculations and areflexia. Figure 1 outlines these features.
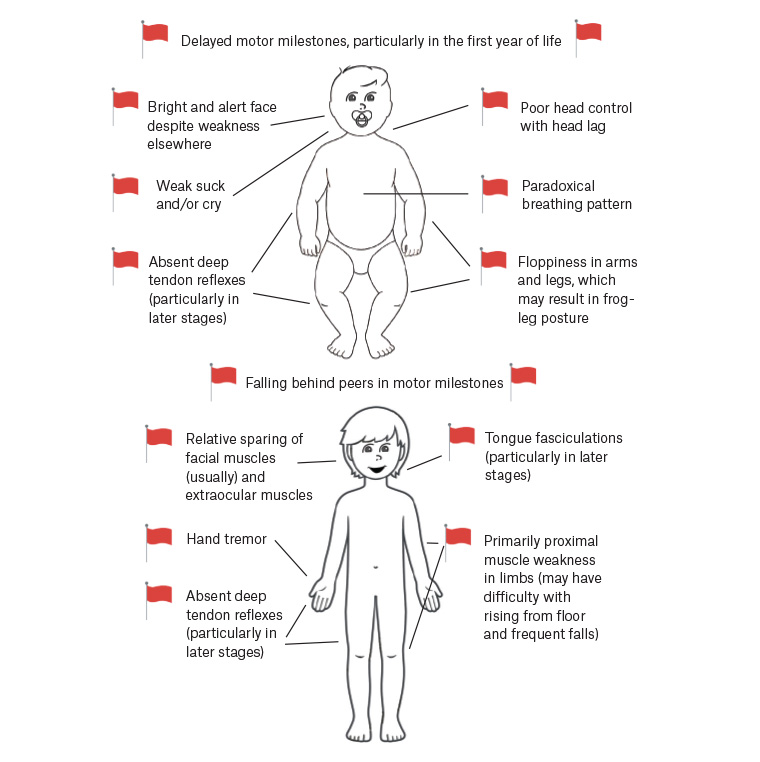
Figure 1. The typical clinical features of an infant and child with spinal muscular atrophy
Many of these symptoms and signs have overlap with other neuromuscular conditions, such as congenital myopathies, limb–girdle muscular dystrophies and dermatomyositis. Suspicion for SMA is important, as it can be delineated on testing, allowing for implementation of disease-specific treatment.
How to refer a child with worrying signs or symptoms
Possible cases of SMA type 1 are best referred promptly and directly to the neuromuscular clinic in each state via the local tertiary paediatric hospital. If the practitioner is working in a regional centre without a tertiary paediatric centre in reasonable proximity, it may be best to refer initially to the local paediatric service, noting concern regarding possible SMA to ensure the referral is triaged appropriately. A single blood test can confirm if a child has the mutations in the SMN1 gene that causes SMA. Practitioners working in remote regions who have clinical concern regarding SMA can arrange this test to be collected locally and sent to a specialised laboratory, following discussion with a geneticist or neuromuscular specialist.
Diagnostic delays are known to occur in SMA. A systematic review revealed the time between symptom onset and diagnosis was 3.6, 14.3 and 43.6 months for SMA types 1, 2 and 3, respectively.9 Preclinical and clinical studies have collectively shown the benefits of early initiation of treatment in SMA for the most efficacious and optimal outcomes.10
Across several countries internationally and as a pilot program in Western Australia, New South Wales and the Australian Capital Territory, newborn screening for SMA is being introduced to expedite early detection for SMA and allow treatment initiation.11,12 It is important to note, however, that a normal previous newborn screening test, while often reassuring, cannot exclude all cases of SMA as the sensitivity is approximately 95%. Consequently, clinical suspicion for SMA should always result in a timely referral to the local paediatric neuromuscular services.
Prognosis and treatment options
There are now several SMN-dependent disease-modifying therapies for SMA: two SMN2 splice modulators and gene therapy.10,13–16 Table 2 summarises the major treatments, their methods of action, administration details and important side effects or details for GPs to know and commercial status in Australia in October 2021.
| Table 2. A summary of disease-modifying therapies for spinal muscular atrophy10,13–16 |
| Therapy |
TGA indication and PBS reimbursement in Australia |
Mechanism of action |
Administration |
Safety information |
Nusinersen
|
Patients with SMA types 1–3 initiating treatment before 18 years of age.
Presymptomatic SMA with two SMN2 copies. |
Antisense oligonucleotide that targets an intronic splicing silencer and thus promotes exon 7 inclusion in SMN2 pre-mRNA to encode full-length SMN protein. |
Intrathecal with four loading injections in first two months then ongoing maintenance every four months. |
Reported side effects largely relate to mode of administration and may include significant treatment anxiety, localised pain and post–lumbar puncture headache. |
Onasemnogene abeparvovec (previously known as AVXS-101)
|
Presymptomatic and symptomatic SMA with 1–3 copies of SMN2 aged less than nine months.
PBAC recommendation of Section 100 (Highly Specialised Drugs Program) listing at September 2021 meeting.
An outcomes-based Risk Sharing Arrangement is required before possible PBS reimbursement. |
SMN1 gene replacement therapy, using an adeno-associated virus capsid to supply a functional SMN1 gene to motor neuron cells. |
Single dose of virus vector given intravenously. |
Liver enzymes may become elevated and cause acute serious liver injury.
Decreased platelet counts could occur following infusion and can result in thrombotic microangiopathy.
Patients will receive an oral corticosteroid before and after infusion and will undergo regular blood tests to monitor liver function. |
Risdiplam (originally known as RO703406)
|
PBS listed for patients with SMA types 1–3 initiating treatment between the ages of two months and 18 years. |
Small molecule that promotes exon 7 inclusion in SMN2 pre-mRNA and full-length SMN protein production. |
Oral medication with good bioavailability in the CNS and peripheral tissues. |
No serious adverse events related to the medication have been reported in studies to date. |
| CNS, central nervous system; PBAC, Pharmaceutical Benefits Advisory Committee; PBS, Pharmaceutical Benefits Scheme; SMA, spinal muscular atrophy; SMN, survival motor neuron; TGA, Therapeutic Goods Administration |
Clinical trials and real-world evidence in people with SMA (defined by age, SMA type and baseline motor function) have shown that these therapies produce improvements in survival, motor function and quality of life. The effects of treatment for all people with SMA are yet to be fully determined; however, it is expected that treatments will provide stabilisation and modest improvements in current motor function rather than recovery of already lost motor neurons.10,13,14 Initiation of treatment in the presymptomatic phase for infants predicted to develop SMA in infancy or childhood has shown the greatest efficacy, with 88% achieving independent walking at a mean age of 20.4 months (range 15.5–29.7 months) and none requiring permanent ventilatory support.17
Multidisciplinary management approaches for SMA continue to form a pivotal part of treatment. Even with treatment, affected patients are likely to experience complications and ongoing disability from SMA. Management goals are individualised, based on the patient’s current motor function. Multidisciplinary care should include specialist neurology, respiratory and orthopaedic care with trained medical professionals, rehabilitation, physiotherapy, nutrition and genetic counselling with active and directed management of comorbidities.18,19 Palliative care remains an important cornerstone in some individuals’ care, and goals are largely determined by the individual patient and family and may involve a combination of novel therapies, non-invasive nutritional and respiratory support.19 It is foreseeable that the GP will play an increasingly active part in the community care of children with SMA treated with life-prolonging disease-modifying therapies, including transition to adult care and/or primarily palliative services. As a key support, the GP typically works closely with the paediatrician and neurology services to optimise health through monitoring growth and nutrition, immunisation and implementation of sick-day plans specific to the individual’s symptoms and health status (eg early initiation of antibiotics, intensification of chest physiotherapy and assessing the need for hospitalisation in respiratory tract infections).18,19 This is summarised in Figure 2.
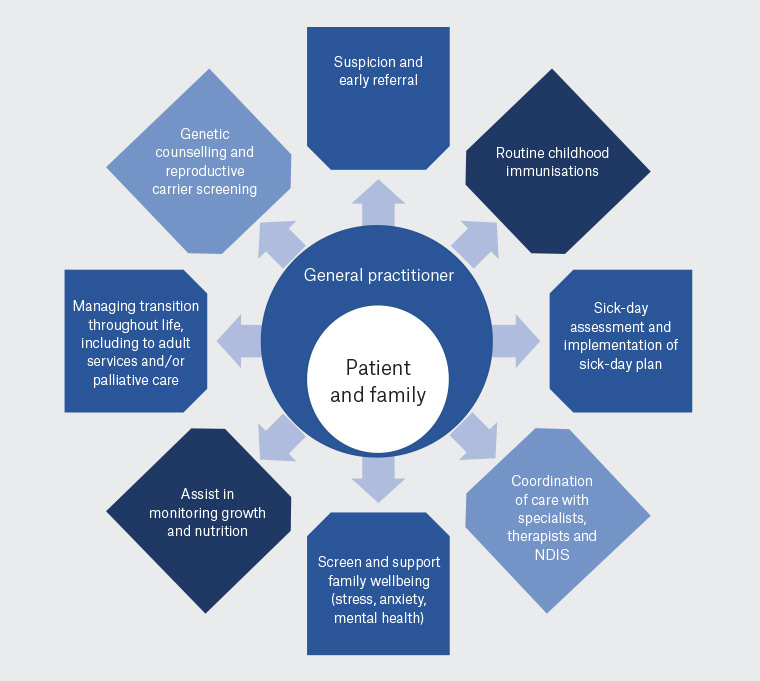
Figure 2. Community management of a child with spinal muscular atrophy overlaps with several important roles that the general practitioner typically provides to families, working in alignment with the multidisciplinary team.17,18
NDIS, National Disability Insurance Scheme
Can spinal muscular atrophy carrier status be screened for?
National guidelines now recommend all women or couples planning a family, or who are in the first trimester of pregnancy, be given the option of genetic carrier screening to understand their risk and allow couples to explore their reproductive choices.20,21 The ethical considerations arising from developments in SMA testing and treatment warrant ongoing examination.
Conclusion
SMA is now a treatable neurodegenerative disease, and early detection and treatment are crucial. As a result of the availability of novel, time-critical treatment options, GPs are now playing a key part in the early recognition of this condition. It is important that all GPs consider urgent referral for a child with delayed motor milestones and signs of symmetrical weakness.
Key points
- SMA is an important cause of childhood morbidity and mortality.
- New disease-modifying therapies, particularly if implemented early, are significantly changing the prognostic trajectory of SMA.
- GPs have a key role in the early detection and referral of these patients, with early suspicion being paramount.
- Key clinical findings are a progressive primary proximal muscle weakness, which in infants includes failure to obtain basic motor milestones in the first year of life, poor head control and floppy tone and posture in an otherwise bright and alert child.
- Affected children may have difficulty keeping up with peers in motor milestones, fall frequently and have difficulty with stairs.
Resources
- ClinicalTrials.gov (www.clinicaltrials.gov) – a database of privately and publicly funded clinical studies conducted around the world. This is a useful site to refer families to for up-to-date information regarding novel treatments.
- Australian Neuromuscular Disease Registry (www.australiannmdregistry.org.au/type-of-diseases/sma) – an Australia-wide registry of people diagnosed with a neuromuscular disease. It collects important medical information from adult and child patients across the country to improve the understanding of neuromuscular disease and accelerate the development of new therapies.