Several skin manifestations have been described in the presentation of, association with and progression of various rheumatic diseases. In addition to their medical implications, these skin manifestations can be a source of patient distress as a result of symptomatology and cosmesis.1 Cutaneous features may precede, co-exist with or follow development of the rheumatic disease. Cutaneous manifestations may allow diagnosis of rheumatological disorders.
The aim of this article is to provide a review of the cutaneous features of five common rheumatological conditions: lupus erythematosus, rheumatoid arthritis, psoriatic arthritis, dermatomyositis and scleroderma.
Lupus erythematosus
Lupus is an autoimmune connective tissue disease classified into systemic lupus erythematosus (SLE) and cutaneous lupus erythematosus (CLE). Clinical disease occurs due to autoantibody formation and immune complex deposition, which lead to pro-inflammatory cytokine production and organ damage. SLE can occur in the absence of cutaneous features, and CLE can occur without systemic disease. Clinical features vary, and various cutaneous manifestations may be seen.2
The skin manifestations may be classified into subtypes. Their key features are outlined in Table 1.1–4
| Table 1. Cutaneous manifestations of lupus erythematosus |
| Subtype |
Clinical presentation |
Systemic lupus erythematosus association |
Scarring |
| Acute |
- Malar (‘butterfly’) rash
- Photosensitive erythema
- Upper limb erythematous papules/plaques
- Cheilitis
- Vasculitis
|
Almost all |
Nil |
| Subacute (Figure 1) |
- Papulosquamous and/or annular rash over trunk and arms
- Photosensitive erythema
|
50% |
Uncommon |
| Chronic (discoid, hypertrophic, mucosal) |
- Localised erythrosquamous patch or plaque with follicular plugging over sun-exposed areas
- Usually on the face and scalp (alopecia)
- Lupus profundus (subcutaneous nodules, followed by lipoatrophy)
- Chilblain lupus (tender, bluish plaques and nodules in cold‑exposed areas)
|
5–10% |
Frequent |
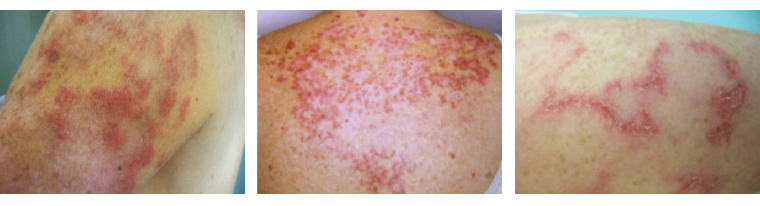 Figure 1. Subacute cutaneous lupus erythematosus
Reproduced from DermNet NZ under Creative Commons non-commercial licensing (https://creativecommons.org/licenses/by-nc-nd/3.0/nz/legalcode).
Figure 1. Subacute cutaneous lupus erythematosus
Reproduced from DermNet NZ under Creative Commons non-commercial licensing (https://creativecommons.org/licenses/by-nc-nd/3.0/nz/legalcode).
Other dermatological associations of lupus include alopecia (as a primary skin manifestation or secondary to CLE), oronasal ulcers, nail dystrophy, digital ulcers, vasculitis and livedo reticularis.5
Key triggers include ultraviolet (UV) exposure and smoking, although various infections and environmental pollutants are also implicated. Smoking increases the risk of therapeutic resistance and refractory disease. Females and those of African and Hispanic descent are at increased risk when compared with the general population.2,5–7 A wide range of medications may trigger drug-induced lupus. Procainamide, hydralazine, quinidine and minocycline are commonly implicated in acute CLE. Terbinafine, proton pump inhibitors, anti-epileptics and biologics including tumour necrosis factor alpha (TNF-α) inhibitors are described culprits in subacute CLE. It should be noted that the use of TNF-α inhibitors may result in positive lupus autoantibodies; however, clinical SLE is rare.8
CLE may be associated with underlying systemic disease.9,10 Diagnosis of SLE requires an antinuclear antibody titre of at least 1:80 plus involvement of other SLE-related clinical and immunological domains, as outlined in the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria (Figure 2).11 This may include cutaneous findings of malar rash, vasculitis, diffuse alopecia and oral ulcers.11

Figure 2. European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for systemic lupus erythematosus
Reproduced with permission of BMJ Publishing Group Ltd from Aringer M, Costenbader K, Daikh D, et al, 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus, Arthritis Rheumatol 2019;71(9):1400–12.
Management of CLE includes sun protection and smoking cessation. Oestrogen-containing medications (ie the oral contraceptive pill, hormone replacement therapy) and sulfonamides may contribute to flares. For localised disease, topical corticosteroids or calcineurin inhibitors (pimecrolimus, tacrolimus) are first-line therapy.12 Widespread or resistant disease may require hydroxychloroquine.12 Doses should remain under 5 mg/kg/day where possible to minimise the risk of retinal toxicity.13,14 For patients taking hydroxychloroquine, baseline screening is required, followed by annual screening after five years for ophthalmological monitoring. Other disease-modifying antirheumatic drugs (DMARDs) may be used as second-line systemic agents.2
Rheumatoid arthritis
Rheumatoid arthritis has specific and non-specific skin manifestations. Rheumatoid nodules, rheumatoid vasculitis and granulomatous dermatoses are key manifestations to consider.2 Associated skin changes are listed in Box 1.15
| Box 1. Cutaneous features associated with rheumatoid arthritis |
- Rheumatoid nodules
- Rheumatoid vasculitis
- Accelerated rheumatoid nodulosis
- Interstitial granulomatous dermatitis
- Rheumatoid neutrophili dermatoses or Sweet syndrome
- Pyoderma gangrenosum
- Livedo racemosa
- Palmar erythema
|
The most common finding in patients with rheumatoid arthritis is rheumatoid nodules.2 These typically present as skin-coloured, firm, painless subcutaneous nodules of varying size over the extensor surface of the peripheral limbs and trauma-related areas. Internal organs, such as the lungs, can also be involved. Histopathology is distinctive, with granulomatous tissue changes within the dermis with surrounding histiocytes, lymphocytes and giant cells.16 Rheumatoid nodules are closely associated with severe disease, rheumatoid factor positivity and titre, and the HLA-DR4 gene.2,15 They do not suggest poorer prognosis. Treatment is rarely indicated, but intralesional corticosteroids or surgical excision can be advised. Lesions may progress to infection or ulceration.15
Rheumatoid-associated vasculitis is uncommon and can present as petechiae/purpura, digital infarcts, cutaneous ulcers and livedo reticularis.15,17,18 It primarily affects small- to medium-sized vessels and indicates poor prognosis.2 It is mainly seen in patients with longstanding seropositive rheumatoid arthritis and multiple other extracutaneous manifestations.15,19 Leukocytoclastic vasculitis is often seen on skin biopsy.20 Systemic involvement needs exclusion and may manifest as fever, fatigue, weight loss and myalgias.
DMARDs used in rheumatoid arthritis can cause cutaneous side effects, but these are uncommon. Methotrexate may cause oral ulcers and alopecia.Sulfasalazine and leflunomide may result in medication hypersensitivity reactions. Hydroxychloroquine can cause hyperpigmentation. TNF-α inhibitors may result in psoriasiform or lupus-like skin reactions.2
Psoriatic arthritis
Skin psoriasis is a very common but non-essential criterion in the diagnosis of psoriatic arthritis.21 Psoriasis usually has a characteristic appearance of well-defined erythematous scaly plaques. The prevalence of psoriasis is 3%, and anywhere from 6% to 42% of patients with psoriasis will go on to develop psoriatic arthritis. Psoriatic arthritis can co-present with (15–20%) or precede (10–15%) skin involvement, especially in older individuals.21 Symptoms and signs of inflammatory arthritis should be inquired after in patients with psoriasis. Investigations for psoriatic arthritis should only be performed for patients with suspicious clinical features.
Nail psoriasis (ie pitting, onycholysis) and scalp involvement have strong correlations with psoriatic arthritis (Figure 3). Psoriatic arthritis is closely associated with the HLA-B27 gene. There is no correlation between the severity of skin psoriasis and the likelihood of developing arthritis.2,21
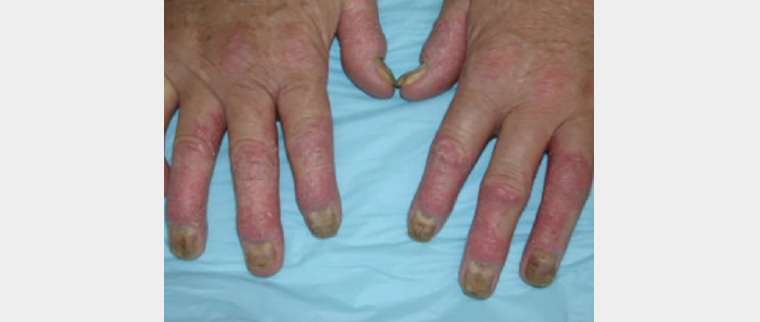
Figure 3. Psoriatic arthritis of the hands showing nail dystrophy and skin psoriasis
Reproduced from DermNet NZ under Creative Commons non-commercial licensing (https://creativecommons.org/licenses/by-nc-nd/3.0/nz/legalcode).
Optimal interventions should target cutaneous and musculoskeletal features. Conventional DMARDs such as methotrexate, sulfasalazine and leflunomide have been the predominant therapy in psoriatic disease. When indicated, biologics (particularly TNF-α blockers, interleukin [IL]-23 inhibitors and IL-17 inhibitors) are efficacious in the treatment of both skin and joint disease.21
Dermatomyositis
Dermatomyositis is an idiopathic microangiopathy characterised by skin changes with (or without, in some cases) myopathy. In myopathic cases, the rash precedes muscle weakness in up to 50% of patients. The pathognomonic features of dermatomyositis include Gottron’s sign, an erythrosquamous rash over the extensor finger joints; Gottron’s papules, which may also occur over other bony prominences; and heliotrope rash, appearing as periorbital erythematous macules.2,22 The pattern of hand involvement can allow dermatomyositis to be distinguished from CLE, which tends to spare the interphalangeal joints (Figure 4). Other commonly seen features include periungal telangiectasia, ragged cuticles, violaceous macules over the neck and upper trunk (Shawl sign) and a photosensitive V-neck rash.22 Less commonly, facial erythema, psoriasiform changes, panniculitis (typically painful nodules or plaques with overlying erythema) or alopecia may be seen. Mechanic’s hands, characterised by well-demarcated hyperkeratotic erythematous changes, are frequently seen in antisynthetase syndrome. This has prognostic significance, as patients with this condition are at high risk of developing interstitial lung disease. Calcinosis cutis is a common feature of juvenile dermatomyositis.22 Extracutaneous features other than proximal muscle weakness may include fatigue, arthralgia, dysphonia, dysphagia and respiratory muscle weakness.
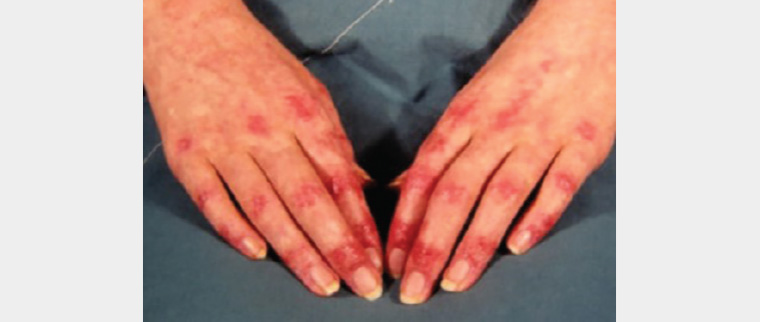
Figure 4. Dermatomyositis of the hands showing Gottron’s papules
Reproduced from DermNet NZ under Creative Commons non-commercial licensing (https://creativecommons.org/licenses/by-nc-nd/3.0/nz/legalcode).
Skin biopsy is useful for diagnosis of dermatomyositis, particularly in amyopathic presentations. A 3 mm punch biopsy should be used. However, the histological features, including interface dermatitis, are usually indistinguishable from CLE.23 Careful clinical correlation is needed for diagnosis. Serology may reveal anti-Mi-2 antibody positivity, and less commonly anti-T1F1 or anti-NXP2 antibodies, which are closely associated with internal malignancy.22,24
The aetiology of dermatomyositis is still largely unknown. There is an established increase in the risk of malignancy in adult patients, particularly those who are amyopathic.25 Therefore, thorough malignancy screening should be performed at the time of diagnosis of dermatomyositis. Malignancy can precede, coincide with or follow the diagnosis of dermatomyositis. Ovarian, lung and gastrointestinal cancers and non-Hodgkin’s lymphoma are the most common.25 Other associations include autoimmune connective tissue disease, cardiovascular disease and interstitial lung disease.2,22 Photosensitivity and mechanical stress appear to be exacerbants.22 Hydroxyurea, TNF-α blockers and IFN-β-1 agents have been described as pharmacological triggers. Medication-induced dermatomyositis may present amyopathically.26
Systemic corticosteroids, and sometimes DMARDs, are used to control cutaneous symptoms.22,26 Management also requires investigation and treatment of any underlying systemic involvement and associations.26 These patients should be monitored closely on subsequent reviews, and it is important to ensure all routine age-appropriate malignant screening is up to date.
Scleroderma
Scleroderma is a term used to describe both localised scleroderma (ie morphea) and systemic sclerosis. Scleroderma is caused by autoantibody deposition, excess collagen production and microvasculopathies.27–29 Localised scleroderma, or morphea, manifests as focal inflammation and sclerosis. It can be limited, linear, generalised or mixed. Morphea occurs in the absence of systemic disease.
Systemic sclerosis is a complex multisystem disorder classified into two major subsets, limited (previously termed CREST [calcinosis, Raynaud’s phenomenon, oesophageal dysmotility, sclerodactyly and telangiectasia] syndrome) and diffuse systemic sclerosis. It is classified largely on the basis of the extent and acuity of the skin changes.27 Although skin changes occur in both forms, faster onset is seen in diffuse sclerosis when compared with limited sclerosis. Additionally, patients with diffuse scleroderma are at high risk of early onset internal organ involvement, including interstitial lung disease and renal crisis.28 Cutaneous changes are characterised by skin tightening, finger swelling, contractures, telangiectasia and Raynaud’s phenomenon (Figure 5). In limited scleroderma, skin sclerosis tends to affect the face, neck and distal limbs, whereas diffuse scleroderma is more widespread and tends to affect the torso and proximal limbs.27,29 Telangiectasia commonly occurs on the face. The severity of skin sclerosis has been directly correlated with poorer overall prognosis.30 The modified Rodnan skin score, based on the sclerosis severity, is widely used to determine the potential risk of systemic involvement and response to therapy.27,31,32 Significant change in physical appearance can affect patients both functionally and psychologically. Diagnosis can be challenging when scleroderma coexists with another rheumatic disease such as lupus or dermatomyositis; this is known as scleroderma overlap syndrome.
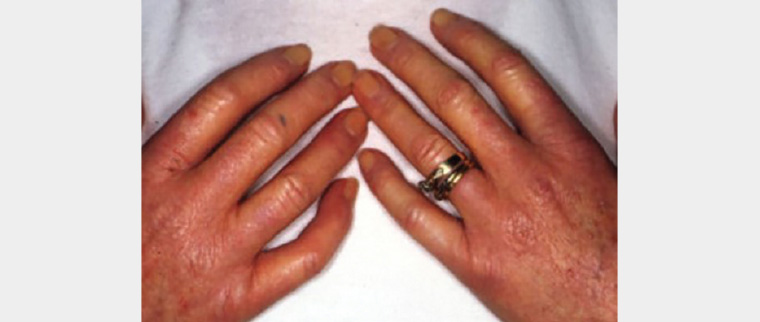
Figure 5. Skin tightening and sclerodactyly of the hands
Reproduced from DermNet NZ under Creative Commons non-commercial licensing (https://creativecommons.org/licenses/by-nc-nd/3.0/nz/legalcode).
Diagnostic features of systemic sclerosis can be seen histologically, typically appearing as marked dermal thickening and dense collagen bundles.33 However, skin biopsy is not required to reach this diagnosis. Serologically, anti-centromere and anti-Scl-70 antibodies are significantly associated with limited scleroderma and diffuse scleroderma, respectively, but may be negative in these conditions. RNA polymerase III has been closely associated with diffuse cutaneous disease in patients with systemic sclerosis.27 In patients with morphea, nailfold capillaroscopy and serological testing should be considered to exclude systemic involvement.
Options remain limited in the treatment of cutaneous sclerosis. Variable response rates have been reported with DMARD therapies.2 UVA phototherapy has shown benefit in some cases.34 More recently, autologous stem cell transplantation has emerged as an alternative with promising results from clinical studies to date.35 Digital ulcerations secondary to Raynaud’s phenomenon may be successfully treated with vasodilator therapy, which can include iloprost infusion. For localised morphea, there are various therapies available ranging from topical or intralesional corticosteroids to methotrexate for active deep lesions, but this condition is generally self-limiting over approximately five years.36
Conclusion
Comprehensive assessment of the cutaneous findings of rheumatic conditions is important to enable accurate diagnosis. These features can be particularly debilitating symptomatically, functionally and psychologically. In many cases, patients should receive multidisciplinary input, which may include collaborative management between general practitioners, rheumatologists, dermatologists and allied health professionals. Recognising the implications of rheumatic skin disease is crucial to enable prompt and holistic management of these complex heterogeneous diseases.
Key points
- Cutaneous manifestations of rheumatological disease are variable and have implications in diagnosis, prognosis and treatment.
- CLE may occur without systemic disease.
- Nodules, vasculitis and granulomatous dermatoses are the most common cutaneous features of rheumatoid arthritis.
- Psoriatic arthritis can co-exist with, follow or precede skin psoriasis.
- Systemic sclerosis manifests with significant cutaneous features, which can substantially affect the patient both functionally and psychologically.
- It is critical that clinicians exclude malignancy in adult patients with dermatomyositis.